
Abstract
The pathogenesis of many diseases is most closely connected with aberrantly regulated apoptotic cell death. The past 15 years have witnessed an explosion in the basic knowledge of mechanisms that regulate apoptosis and the mediators that either trigger or inhibit cell death. Consequently, great interest has emerged in devising therapeutic strategies for modulating the key molecules of life-and-death decisions. Numerous novel approaches are currently being followed employing gene therapy and antisense strategies, recombinant biologics or classical organic and combinatorial chemistry in order to target specific apoptotic regulators. Although drug development is still in its infancy, several therapeutics have progressed to clinical testing or have even been approved in record time. This review outlines the recent advances in the field of apoptosis-based therapies and explores some highlights of a very active field of drug development.
Similar content being viewed by others
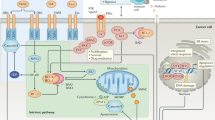
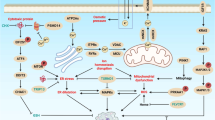
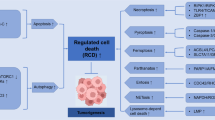
Introduction
The discovery of apoptosis was in some respect exceptional for basic research, since the potential of its application for therapy of various human diseases became almost immediately evident. The human body is composed of approximately 1014 cells. Every day billions of cells die an altruistic death in order to secure the functionality of the whole organism. Cell death like this is essential for the regulation of development, and later on for maintenance of tissue homeostasis, being a consequence of a balanced cell death versus cell proliferation ratio. Equally, or perhaps even more important, is the role of apoptosis as a cause of disease. Dysregulation of apoptosis can result in severe pathological syndromes. Acute pathologies such as stroke, heart attack or liver failure are associated with life-threatening sudden death of whole tissue areas or organs, whereas certain neurodegenerative syndromes are the result of a more slowly progressing cell death. Cancer, in contrast, can be caused in part by a prolonged lifespan of transformed cells that would be normally eliminated. A reasonable estimate is that either too little or too much cell death contributes to approximately half of the medical illnesses, for many of which an adequate therapy is lacking.
To date, most of the key players in cellular apoptosis regulation are identified and can be targeted by therapeutic strategies. These include death receptors triggering apoptosis from the cell surface, Bcl-2 proteins as the gatekeepers of the mitochondrial pathway, caspases as the executioner enzymes or endogenous caspase inhibitors. The identification of the major regulators of apoptosis has boosted an intense research in developing therapeutic approaches to intervene with cell death, either in a pro- or antiapoptotic direction. For instance, extracellular death signals sensed by death receptors can be mimicked by recombinant ligands or agonistic antibodies. Caspases can be either inhibited or activated by small-molecule drugs. Undesirable proteins, like prosurvival factors in cancer cells, can be specifically downregulated by antisense or other strategies. Finally, important protein–protein interactions can be interfered with or mimicked by peptides or organic compounds.
During the last decade, substantial advances have been achieved in the field of apoptosis-based therapeutics. First-generation attempts mainly used inefficient gene delivery approaches, recombinant proteins and peptides, or relatively nonselective drugs that were of limited use for clinical application. Nevertheless, these preclinical experiments, performed in cell culture and animal models, provided the important proof-of-principle evidence that targeting of apoptosis is a valid strategy for a large number of diseases.
Currently, a great deal of effort is being aimed at reducing the peptidic nature of prototypic drugs and at replacing them by small-molecule organic compounds, which could set the stage for future therapeutics. Improved oligonucleotide chemistry has expanded the potential use of antisense constructs by enhancing their stability and lowering associated toxic side effects. Furthermore, advances in combinatorial chemistry led to the rapid assembly of chemical libraries containing vast numbers of drug derivatives. Combined with innovative library screening techniques, structural biology and bioinformatics, this has greatly accelerated the development of lead compounds that could progress into clinically applicable drugs. Apoptosis-targeting therapies are now advancing from preclinical/clinical trials to actual application. This is a remarkable success regarding the short time period since the discovery of apoptotic cell death.
Therapeutic Approaches Targeting Death Receptors
A major pathway of apoptosis is mediated by cellular death receptors belonging to the tumor necrosis factor (TNF) receptor superfamily.1 In a simplified model, the activation of a surface death receptor by its cognate death ligand, TNF-α, CD95 ligand (CD95L) or TNF-related apoptosis-inducing ligand (TRAIL), leads to the intracellular recruitment of the death-inducing signaling complex (DISC) by means of protein/protein interactions involving death domains. As recently reported, composition and signaling of the TNF receptor DISC differ considerably from the CD95 or TRAIL receptor-bound DISC.2 Nevertheless, common to all DISCs is the recruitment of caspase-8 and probably caspase-10 resulting in the activation of the initiator caspase and the subsequent cleavage-mediated activation of downstream effector caspases. An exciting finding with respect to a potential therapeutic application was the observation that death ligands like TNF or CD95L could efficiently induce cell death of a variety of tumor cells. However, despite most promising prospects, cancer therapy involving the TNF or CD95 receptor/ligand systems mostly failed, owing to severe systemic toxicity of TNF and CD95 agonists in mice and humans.3 These findings raised a general scepticism about the feasibility of cancer therapy involving death ligands without accompanying severe, especially hepatotoxic side effects.
However, the discovery of TRAIL raised new hopes. TRAIL seemed to be a most promising candidate for cancer therapy, because it specifically killed tumor cells while leaving normal tissues relatively unharmed. Injection of TRAIL into athymic SCID mice challenged with human mammary adenocarcinoma, colon carcinoma or cholangiocarcinoma induced tumor cell apoptosis, suppressed tumor growth and, moreover, improved survival substantially.4, 5 TRAIL was not only able to prevent tumor growth of freshly xenotransplanted tumors, but, more importantly, decreased the size of established tumors. Furthermore, preclinical safety studies in non-human primates demonstrated no toxicity of TRAIL treatment, even when doses up to 10 mg/kg/day were applied.6
Concerns regarding the safety of applying TRAIL to humans arose, when polyhistidine-tagged recombinant TRAIL was found to induce apoptosis of cultured human primary hepatocytes.7 Similarly, death of human brain cells was inflicted by trimerized Flag-tagged TRAIL.8 To date, the cause of these findings seems to be clarified as being due to the preparation and tag version of TRAIL, which alters its biochemical properties decisively. After replacement of histidine-tagged TRAIL by the nontagged protein, toxicity in human or cynomolgus monkey hepatocytes could no longer be observed.9
Most current cancer treatments like chemo- or radiotherapy engage the intrinsic mitochondrial pathway to initiate cell death. Apoptosis resistance of tumors is often caused by inactivation of integral parts of this pathway, for example, through mutation of the tumor suppressor p53 or overexpression of antiapoptotic Bcl-2 family members. TRAIL-mediated apoptosis, in contrast, is independent of p53 and can bypass overexpression of Bcl-2-like proteins by directly activating the caspase cascade. Indeed, combination treatment of TRAIL with conventional chemotherapeutic agents or ionizing radiation mostly results in strongly enhanced cytotoxic effects. Chemo- and radiotherapy can even sensitize previously nonresponding tumor cells to TRAIL. The observed synergistic effects are not only due to the activation of both the intrinsic and the extrinsic apoptotic pathways, but also other cellular mechanisms potentiate this effect, such as transcriptional induction of the TRAIL receptors R1 and R2, a reduced expression of antiapoptotic proteins such as Bcl-2 or Bcl-xL and an upregulation of proapoptotic molecules such as caspase-8 and FADD.10
Currently, there are several clinical and preclinical studies underway investigating the therapeutic potential and safety of TRAIL agonists as anticancer agents (Table 1). Genentech and Amgen plan to start clinical phase 1 trials with soluble TRAIL this year. An agonistic TRAIL-R2 antibody (TRA-8) was generated by Sankyo Co., Ltd. and proved to be cytotoxic against primary hepatocellular carcinoma cells without inducing cell death in normal hepatocytes.11 Already now, several antibodies against TRAIL-R1 and -R2, developed by Human Genome Sciences Inc. and Cambridge Antibody Technologies (CAT), respectively, are investigated in phase 1 and 2 clinical trials. So far, treatment of patients with advanced solid tumors or non-Hodgkin's lymphoma revealed no hematological or hepatic toxicity of the TRAIL-R1-specific antibody HGS-ETR1 with doses of up to 10 mg/kg of body weight. Six of 57 patients enrolled in a preliminary trial even reached stable disease.
Because of the superior tumor killing activity of TRAIL agonists and the toxic side effects associated with systemic treatment of other death ligands, TNF had temporarily sunken into oblivion. But besides its antitumor effect, TNF efficiently destroys tumor-supplying blood vessels by apoptosis and, furthermore, improves the permeability of the vasculature to cytotoxic drugs. Based on these findings, Lejeune and colleagues developed a strategy to treat locally advanced melanomas and sarcomas: high-dose TNF treatment combined with chemotherapeutic drugs was applied by isolated limb perfusion resulting in complete response rates in patients.12 TNF was meanwhile demonstrated to considerably improve penetration of melphalan and doxorubicin into tumors in animal models. Interestingly, TNF specifically disrupted tumor-associated blood vessels, while leaving normal tissues and blood vessels unharmed. Curnis et al.13 have recently exploited this increase of drug penetration induced by very low doses of TNF for the treatment of melanoma and lymphoma with doxorubicin. To target TNF to the vasculature, TNF was coupled to the GNGRC peptide, a ligand of aminopeptidase N (CD13) that binds specifically to endothelial cells. Mice with established xenograft lymphomas could be cured by this approach, demonstrating that targeted delivery of TNF may considerably enhance its therapeutic properties.
TNF is involved in a wide spectrum of biological pathways and, moreover, is the key mediator of inflammatory responses. Therefore, not surprisingly, dysregulation of TNF responses causes severe pathological syndromes including rheumatoid arthritis and Crohn's disease. Animals with inflammatory bowel disease benefit significantly from treatment with anti-TNF antibodies or genetic TNF knockout.14 Symptoms in about 60% of patients improved considerably upon single infusions with a chimeric monoclonal anti-TNF antibody (infliximab, remicade).15 Infliximab (Centocor, Schering-Plough Corporation) comprises the variable region of a mouse anti-human TNF antibody fused to the constant region of human IgG1. It binds and neutralizes TNF, both soluble and membrane-bound forms, and lyses cells expressing membrane TNF. CDP571 (humicade), another humanized anti-TNF antibody developed by Celltech and Biogen, works similar. While infliximab is already approved for the treatment of rheumatoid arthritis and inflammatory bowel disease, CDP571 has entered phase III clinical trials, but with so far disappointing results for Crohn's disease.16 CAT and Abbott developed a next-generation recombinant anti-TNF antibody (adalimumab, Humira), in contrast to the murine chimeric proteins infliximab and CDP571. Subsets of patients treated with infliximab experienced allergic reactions. The fully human antibody adalimumab might be a suitable substitute treatment for those patients with acute hypersensitivity reactions to chimeric antibodies.17 Adalimumab is approved for the treatment of rheumatoid arthritis, and owing to excellent results in phase 3 trials will be presumably approved for psoriasis soon. Another clinically relevant anti-TNF strategy was developed by Immunex: Etanercept (Enbrel) is a chimeric TNF-inhibitory molecule composed of the extracellular TNF-binding region of TNF receptor 2 fused to an IgG heavy chain.18, 19 Etanercept, marketed by Amgen and Wyeth, is already approved for the treatment of rheumatoid arthritis, psoriasis, spinal arthritis and Crohn's disease.
Also the CD95 receptor/ligand system is most likely a very appealing target for therapeutic intervention. A most devastating acute trauma is the injury of the spinal cord. Initial studies on spinal cord injury demonstrated that inhibition of apoptosis by caspase inhibitors was beneficial for the regeneration process.20 In a recent study, Demjen et al.21 together with Apogenix Biotechnologies showed that treatment of mice experiencing spinal cord injury with neutralizing antibodies to CD95L effectively decreases cell death at the lesion site. Furthermore, only several weeks after injury, mice treated (prior to transection of the spinal cord) with anti-CD95L-antibodies could again actively move their hind legs. Recovery was associated with regenerating corticospinal fibers.
CD95/CD95L have been implicated in further severe syndromes: stroke, multiple sclerosis (MS), graft-versus-host disease (GVHD), toxic epidermal necrolysis (TEN) and others. For instance, CD95- and TNF-mediated signaling has been implicated in stroke-associated tissue damage, characterized by massive neuronal death after brain ischemia. Mice deficient in CD95 (lpr mice) or CD95L (gld mice) are partly protected from stroke, and even more so if TNF is inactivated in addition. Injection of mice with anti-CD95 and anti-TNF antibodies 30 min after ischemic injury resulted in a marked decrease of both infarct volume and mortality.22 MS is a progressive inflammatory disease caused by selective death of oligodendrocytes, resulting in demyelination of motoric nerve fibers. Several experimental findings hinted at a possible involvement of CD95 in MS, as patients revealed elevated soluble CD95 levels in cerebrospinal fluids together with a markedly increased expression of CD95 in MS lesions.23, 24 GVHD is a complication associated with bone marrow transplantation from allogeneic donors. The transplanted donor lymphocytes attack host organs, especially skin, gut and liver. In its mild form, this can be useful for the eradication of persistent leukemia; however, aggressive GVHD can severely endanger the patient's life. In animal models, anti-CD95L antibodies could inhibit GVHD.25 It was also demonstrated that, even though inactivation of TNF or perforin could reduce GVHD, only targeting of CD95 did not ablate the beneficial graft-versus-leukemia effect.26 TEN is a severe and often fatal reaction to drugs, characterized by massive keratinocyte apoptosis resulting in the detachment of large areas of epidermis. Highly increased expression of functional CD95L has been observed in CD95-positive TEN keratinocytes. Progression of TEN in patients could be blocked by administration of antibodies interfering with CD95/CD95L interaction.27
Because death receptor pathways are involved in a wide variety of serious human syndromes, for which curative therapies are not available, they are most interesting targets for the development of new, alternative therapeutic regimens. Nevertheless, disadvantages associated with protein-based therapies clearly exist: proteins have to be administered systemically, in high doses, and for chronic diseases regularly. In this regard, nonpeptidic agents such as caspase inhibitors might turn out as the superior treatment for at least certain apoptosis-associated diseases.
Pharmacological Approaches of Caspase Inhibition
Caspases are a family of cysteinyl aspartate-specific proteases comprising 12 human members. They recognize tetrapeptide motives (P4–P1) in their substrates and require an aspartic acid residue at P1.28, 29 Caspases can be separated into two groups according to their biological function. The first caspase to be identified, the interleukin-1β-converting enzyme (ICE, caspase-1), is crucially involved in cytokine maturation and inflammatory processes. Most other caspases play key roles in programmed cell death, some of them acting as initiator (caspase-8, -9 and -10) and others as executioner caspases (caspase-3, -6 and -7).
The design of potent caspase inhibitors is a main focus of interest in apoptosis-based drug development. The enormous potential of caspase inhibitors has been demonstrated with prototype inhibitors in several animal models. Liver diseases like alcoholic liver disease or hepatitis B and C virus infection are associated with accelerated apoptosis. In animal models, the broad irreversible caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (z-VAD-fmk) was protective and efficiently blocked death receptor-mediated liver injury.30, 31 In arthritis models, repression of proinflammatory cytokine release (IL-1β, IL-18) by blocking its caspase-1-dependent maturation led to efficient reduction of disease severity.32, 33 Myocardial infarction and the resulting death of myocytes could be ameliorated by z-VAD-fmk and related peptide inhibitors in animal models.34 Also sepsis that is associated with massive apoptosis of lymphocytes and lethal in approximately 29% of human cases was efficiently reduced in a mouse model by z-VAD-fmk, resulting in increased survival.35 Last but not least, after spinal cord injury, activation of extrinsic and intrinsic apoptotic pathways has been demonstrated in animal models, which was efficiently blocked by z-VAD-fmk, leading to reduced lesion size and improved motoric function.36
A typical caspase inhibitor consists of an electrophilic group termed the ‘warhead’, the P1 aspartic acid and the P2–P4 peptidomimetic region. The warhead targets the active cysteine residue of the caspase and depends on its substituents that lead to either reversible (R=aldehyde, nitrile, ketone) or irreversible (R=halo-, acyloxy- or diazo-methylketones) caspase inhibition. The relative advantages of reversible versus irreversible inhibitors as well as of specific versus pan-caspase inhibitors are often debated. Irreversible inhibitors exhibit a greater potential in cellular assays compared to reversible inhibitors; however, they are in some cases less specific and affect also other cellular proteases.37 For instance, the fmk group of the irreversible inhibitor z-VAD-fmk was shown to interact with nonrelated cysteine proteases such as cathepsins.38
To overcome the problems associated with the peptide nature of the first prototype caspase inhibitors, several approaches are being employed: (1) the stepwise reduction of the peptidic nature, (2) the use of compounds mimicking the peptide backbone or (3) the modification of the active warhead. The P1 aspartic acid is crucial for the activity of the inhibitor, and substitution in most cases results in loss of activity. Only a few classes of inhibitors without aspartate at P1 have been reported so far. One example was developed by Okamoto et al.,39 who replaced the P1 aspartate carboxyl group with an acyl-sulfonamide. Other developed inhibitors without an aspartate in P1 position comprise isatin sulfonamides, which are potent reversible, nonpeptidic inhibitors of caspase-3 and -7.40 GlaxoSmithKline further disclosed a series of similar piperidinyl substituted isatins and related 5-alkylaminosulfonyl-3,3-dichloro-oxindoles. Also Pfizer has developed isatin-based caspase inhibitors, from which for instance the compound MMPSI, a 2-methoxymethylpyrrolidinyl-sulfonylisatin, was able to reduce ischemic injury in isolated rabbit hearts or cardiomyocytes with an IC50 of 200 nM.41 AstraZeneca developed a series of nonpeptidic anilinoquinazolines (AQZs).42 Similar to isatin sulfonamides, AZQs contain an electrophilic carbonyl that probably functions as the warhead.43
A critical part of a caspase inhibitor is the active warhead, which not only determines the reversibility of caspase inhibition, but might also cause side effects due to the interference with other cysteine proteases. ICN/Enzyme Systems Products Inc. has recently launched a new broad caspase inhibitor, which is increasingly taken for laboratory use: Q-VD-OPH (quinolyl-Val-Asp(OMe)-[2,6-difluorophenoxy]-methyl-ketone) lacks the often unspecific fmk group, but has increased potency and reduced toxicity. Q-VD-OPH inhibited various caspases with IC50 values of 25–400 nM and was nontoxic in doses up to 1 g/kg after i.p. administration in mice.44
Merck Frosst has designed tetra- and dipeptides in which the active aldehyde warhead was replaced by a γ-keto group. The most potent tetrapeptide inhibitor identified inhibited caspase-3 in vitro efficiently (IC50=48 nM), whereas it was only weakly effective in cellular assays.45 Becker et al.46 therefore reduced the peptide nature of caspase-3 inhibitors by employing a peptidomimetic 5,6,7-tricyclic system or a pyrazinone at P2–P3 and by replacing the negatively charged P4 aspartyl with neutral groups. Recently, Merck also developed nicotinyl aspartyl ketones. The 5-bromonicotinamide derivative of the aspartate aldehyde was a weak inhibitor specific for caspase-3, which could be however improved by conversion of the aldehyde to ketones and further substitution at the 5-position of the pyridine ring.46, 47 Interestingly, the nicotinic acid replacements produced conformational changes in the S2 and S3 subsites of caspase-3 and revealed a previously unrecognized binding region. Several Merck inhibitors are currently in preclinical trials. M-826, a small reversible caspase-3 inhibitor, blocked brain tissue damage in an animal model of hypoxia–ischemia when injected 2 h after ligation.48 In a mouse model of Huntington's disease, M-826 also prevented cell death of striatal neurons.49 The broad-spectrum caspase inhibitor M-920 as well as the caspase-3-specific inhibitor M-791 decreased lymphocyte apoptosis in thymus and spleen of mice subjected to sepsis induced by cecal ligation-puncture and rescued 80–90% of animals from lethal septic shock.50
Whereas the C-terminal warhead is responsible for the reactivity of an inhibitor, N-terminal modifications also alter its effectiveness by influencing cell penetration. Most inhibitors therefore carry a hydrophobic N-protecting group. Cytovia, a Maxim subsidiary, developed a series of dipeptides with an fmk (or similar) warhead and different N-terminal substituents for inhibition of apoptosis and suppression of IL-1β-secretion. The potent, irreversible inhibitor z-VD-fmk, termed MX1013, inhibited several caspases with IC50 values ranging from 5 to 20 nM in vitro and 500 nM in cell assays. z-VD-fmk effectively blocked endotoxemia in a rodent model and, moreover, prevented experimental liver damage caused by CD95 activation at doses of 1 mg/kg i.v. z-VD-fmk also demonstrated neuroprotective effects in a model of transient focal ischemia/reperfusion injury. Cai et al.51 further optimized the N-protecting group of dipeptidic inhibitors. The most potent compound identified in this analysis was MX1122 (2,4-di-Cl-Cbz-Val-Asp-fmk) with a caspase-3 IC50 of 25 nM in vitro and a cell-protecting IC50 of 100 nM. In a mouse model of liver failure, 1 mg/kg of MX1122 administered after injection of anti-CD95 achieved complete rescue of mice.
The P2–P4 peptidomimetic region is the most variable part of the inhibitor and imparts the selectivity for certain caspases. Starting from the structure of the substrate recognition tetrapeptide, inhibitors are often designed by a stepwise substitution with nonpeptidic components. Sunesis has recently developed a novel combinatorial screening method for the identification of suitable P2–P4 backbones, called extended tethering.52, 53 The approach allowed the identification of ligands that bind to discrete regions of caspase-3 and helped to direct the assembly of these ligands into small-molecule inhibitors. First, a small-molecule ‘extender’ that irreversibly alkylates the cysteine residue of caspase-3 and also contains a thiol group was designed. The modified protein was then screened against a library of disulfide-containing small-molecule fragments. Compared to other high-throughput screening (HTS) technologies, this method allowed also weakly interacting compounds to be isolated, which might serve as leads for the development of more efficient inhibitors.54
Already before the discovery of the role of caspases in apoptosis, several companies had started drug programs aimed at screening for caspase-1 inhibitors in order to suppress IL-1β production. Pfizer/Abbott prepared a series of caspase-1 inhibitors and tried to improve its potency by rigidifying the caspase-bound inhibitor through an intramolecular hydrogen bond with an amino moiety of sulfonamides.55 Further substitution of the benzyloxycarbonyl warhead by structurally similar sulfonamide derivatives resulted in an approximately 10-fold reduced caspase-1 IC50, the most potent compound displaying a remarkable caspase-1 IC50 of 3.4 nM. Pralnacasan (VX-740), an orally active caspase-1-specific and reversible inhibitor developed by Vertex for the treatment of rheumatoid arthritis, significantly reduced patient symptoms in phase 2a clinical trials. Unfortunately, pralnacasan induced abnormal liver toxicity upon long-term treatment in animal studies and was recently withdrawn from phase 2b. Further phase 1 trials are therefore underway, and a new-generation inhibitor VX-756 is currently tested for the treatment of inflammatory diseases. Together with Serono, Vertex has developed VX-799, a pan-caspase inhibitor for the treatment of sepsis, which demonstrated efficacy in different models of organ failure.
Pioneering work in the area of caspase inhibitor design has been undertaken by Idun. They developed C-DEVD-H, a conformationally constrained peptidomimetic caspase inhibitor, serving as a lead for the further development of acyl dipeptide compounds.56, 57, 58 Idun recently designed potent caspase inhibitors based on oxamyl dipeptides.59 The first caspase inhibitor to finally enter clinical trials has been IDN-6556, and so far with some success. Preclinical studies with IDN-6556 demonstrated selective and irreversible inhibition of several caspases with low to subnanomolar IC50 in vitro and submicromolar range in vivo. Mouse models of liver injury already indicated that IDN-6556 could be a prime candidate for the treatment of liver diseases, because it efficiently reduced serum levels of liver transaminases, irrespective of the mode of administration. IDN-6556 also exhibited marked post-insult efficacy and blocked lethality completely, even when administered 4 h after anti-CD95 treatment.60 The potency of this inhibitor was also proven in the bile duct ligation model.61 Administration of IDN-6556 significantly reduced hepatocyte apoptosis and, importantly, also liver inflammation and fibrosis.
In a first clinical study, healthy volunteers and patients with impaired hepatic function were treated with i.v. infusions of IDN-6556. All but one patient exhibited significant falls in serum transaminase levels. Doses up to 1.5 mg/kg for 7 days were well tolerated, and moderate side effects were phlebitis and inflammation at the site of infusion.62 IDN-6556 also normalized transaminase levels of hepatitis C virus (HCV)-infected patients, if given orally twice a day for 2 weeks. Orally given IDN-6556 is currently tested in phase 2 clinical trials for the treatment of HCV infection; further studies are planned for nonalcoholic steatohepatitis and other hepatobiliary diseases.
Moreover, it was demonstrated that apoptosis of sinusoidal cells, a major problem in liver transplantation caused by cold ischemia–warm reperfusion liver injury, could be effectively reduced by IDN-6556 in an animal model.63 In contrast to prior experiments with the caspase inhibitor IDN-1965, which absolutely required coadministration to the organ donor the preservation solution as well as to the liver recipient,64 IDN-6556 was effective even when added only to the preservation solution. Therefore, in 2003, the FDA granted orphan drug status to IDN-6556 for the treatment of patients receiving liver transplants. Currently, phase 2 clinical trials are underway involving transplant centers in the US and Europe.
Therapeutic Approaches Leading to Caspase Activation
Besides inhibition of caspases to suppress cell death, also the opposite effect, the activation of the death machinery by caspase activators, might be a valuable tool, especially in the therapy of human cancers. Although more preliminary, several strategies aiming at activation of caspases specifically in tumor cells are currently being designed. Inducible caspases have been engineered by fusing them to chemical dimerization domains. After delivery of these chimeric ‘death switches’ by adenoviral gene transfer, caspases can be activated to trigger apoptosis in tumor cells by cell-permeable dimerization drugs.65, 66 Inducible caspase-9 (iCasp9) under the control of an androgen-responsive promoter was specifically targeted to prostate cancer cells.67 In vivo, iCasp9 induced apoptosis in xenografted prostate tumors, but not in hepatoblastoma after chemical induction. Several related gene therapeutic approaches to deliver caspases have been investigated and confirmed their antitumor activity in both in vitro and in vivo studies. For instance, caspase-6 under control of the human telomerase reverse transcriptase promoter (hTERT) was transferred into glioma cells.68 This approach is assumed to trigger tumor-specific apoptosis, since telomerase expression is reactivated in tumor cells, whereas differentiated cells hardly express the enzyme.
Other chimeric proteins, called immunocasp-3 and immunocasp-6, have been created by fusing a single-chain anti-erbB2/HER2 antibody (e23sFv) to the translocation domain of Pseudomonas exotoxin-A (PEA) and an active caspase.69, 70 Tumor specificity of this strategy is provided by erbB2/HER2, which is overexpressed in 20–40% of a variety of human neoplasms including breast, ovarian, endometrial, gastric, bladder, prostate or lung cancer. The antibody used in this approach binds to the extracellular domain of HER2 and is internalized by endocytosis. The translocation domain of PEA causes the release of the chimeric protein from internalized vesicles and subsequent autoactivation of the caspase. Death-inducing activity of the construct could be demonstrated in HER2-overexpressing tumor cells as well as in a xenograft model. Immunex/Amgen are currently evaluating caspase-3 linked to the HER2-antibody Herceptin (Genentech) as a clinical treatment. An intriguing novel strategy to target human immunodeficiency virus (HIV)-infected cells in AIDS patients was published by Vocero-Akbani et al.71 The caspase maturation sites in procaspase-3 were replaced by HIV protease recognition motifs. Upon transduction of this construct, HIV-infected cells processed procaspase-3, thereby leading to caspase-mediated apoptosis selectively in infected cells.
As the success of gene delivery approaches is still limited, pharmacological activation of cellular caspases by small cell-permeable drugs might provide a more efficient venue to target cancer cells. Procaspase-3 is held in an inactive, dormant state in healthy normal cells by an intramolecular electrostatic mechanism. This so-called ‘safety catch’ consists of a triplet of aspartate residues located within a flexible loop near the junction of the large and the small caspase subunits. In order to activate caspase-3 from the inactive precursor, the junction between the large and small subunits has to be cleaved. Genetic removal of the safety catch loop resulted in increased autocatalytic maturation and susceptibility to caspase-9,72 suggesting that release of the caspase-3 safety catch may be an important determinant of apoptotic competency. Furthermore, cellular acidification, which is typical in early apoptosis, promotes the disruption of electrostatic interactions and results in caspase-3 activation. In this context, screening for specific drugs that disrupt the safety catch might be a promising approach in order to lower the threshold of caspase activation.
That such direct activators of caspases might be found is supported by the observation that RGD peptides can directly bind to and activate caspase-3. Usually, RGD tripeptides are recognized by integrins in extracellular matrix proteins and can block integrin-mediated signaling and cell adhesion. In several cell types, it was previously shown that soluble RGD peptides induce apoptosis, which was first attributed to the loss of cell attachment and survival signaling. However, Buckley et al.73 demonstrated a direct intracellular activation of caspase-3 by RGD peptides, which was independent of integrins. Indeed, caspase-3 has an RGD tripeptide sequence near its active site, which was proposed to keep the enzyme in a quiescent state. RGD peptides might therefore disturb this intramolecular interaction leading to the activation of the protease. RGD peptides are already in clinical use as antithrombotic drugs. Merck in collaboration with the Scripps Research Institute developed a promising RGD-based candidate drug, inhibiting the new formation of tumor blood vessels. Thus, caspase-activating properties would clearly add to the therapeutic potential that RGD peptides have already established.
Several institutions and companies are performing HTS assays for compounds that could induce caspase activity either in living cells or in vitro. Jiang et al.74 identified a small-molecule drug, α-(trichloromethyl)-4-pyridineethanol (PETCM), that could activate caspase-3 in cell extracts. PETCM relieved the inhibition of apoptosome formation imposed by the oncoprotein prothymosine-α. Furthermore, downregulation of prothymosine-α by RNA interference sensitized cells to apoptosis induction. However, even though a novel role of prothymosine-α has been identified in this study, PETCM is an unlikely therapeutic agent, as high concentrations (200 μM) are required to achieve caspase-3 activation in vitro. In a similar approach, Nguyen and Wells75 (Sunesis) screened a chemical library for caspase-3-activating or -inhibiting compounds in HeLa cell cytoplasmic extracts supplemented with cytochrome c. Structure–activity relationship (SAR) of the most potent agents led to the identification of dichlorobenzyl carbamates and indolones as strong caspase activators. The activating effect of these compounds turned out to be due to the induction of Apaf-1 oligomerization promoting apoptosome formation. An indolone compound was the most potent in cellular assays activating caspase-3 and cell death with an IC50 of 4–50 μM. At 10 μM, the compound was cytostatic on 40 and cytotoxic on eight cell lines from 48 tumor cell lines of the NCI panel.
Maxim Pharmaceuticals are currently evaluating the potential of a series of small-molecule caspase activators, MX-2060, isolated by a similar HTS approach for caspase-activating drugs in cell-based assays. Maxim's MX-2060 series of caspase-activating compounds are derivatives of gambogic acid, a natural product from the resin of the tree Garcinia hurburyi.76 MX-2167 has been shown to induce apoptosis in multiple cancer cell lines. It suppressed tumor growth up to 90% in a syngeneic prostate animal cancer model. In conclusion, the fact that structurally unrelated compounds, identified in independent screens, converge at the level of apoptosome formation or caspase activation suggests that screening for small-molecule caspase modulators might be a valid approach.
‘Knob’-ing on Heaven's Door: IAPs and SMAC/Diablo
Current knowledge identifies the inhibitor of apoptosis proteins (IAPs) as a major control point in the execution of cell death. IAPs comprise a family of caspase-inhibiting proteins characterized by a shared conserved sequence region, termed the baculoviral IAP repeat (BIR) domain.77 Eight endogenous IAPs are known in the human system, all of which inhibit apoptosis. So far, their main physiological roles seem to be (1) the establishment of a threshold under which caspases are kept at bay and (2) providing a pool of active caspases, which can rapidly execute death after release. Growing evidence also suggests the participation of IAP proteins in other cellular functions apart from inhibiting caspases, including protein degradation, cell cycle control and signal transduction.78
Survivin is an IAP member containing a single BIR domain, which is expressed to high levels in cancer cells, but not in normal cells.79 However, it is still unclear whether this is due to an antiapoptotic role of survivin conferring a survival advantage to the tumor cell or due to other reasons. Survivin is proposed to play a role in cell division,80, 81 and might therefore be expressed at higher levels in actively dividing cancer cells than in normal differentiated cells.
Antisense targeting of XIAP has been shown to sensitize a variety of tumor cell lines to radio- or chemotherapy.82, 83 XIAP antisense therapy combined with vinorelbine also proved to be effective in a mouse xenograft model of lung cancer.84 Oligonucleotides with natural backbones are rapidly degraded by nucleases and are therefore not suitable as therapeutic drugs. In first-generation antisense molecules, one of the oxygens of the phosphate backbone is replaced with sulfur. Those phosphorothioate-modified oligonucleotides have enhanced but yet not optimal stability, they are relatively toxic and not suitable for oral administration. Second-generation oligonucleotides comprise DNA/RNA hybrid backbones with improved pharmacokinetics and reduced toxicity. In collaboration with Hybridon, Aegera developed a second-generation XIAP antisense oligonucleotide, AEG35156/GEM®640. Knock-down of XIAP by AEG35156 demonstrated significant efficacy in preclinical models of multiple cancer types. In March 2004, Aegera initiated phase 1 clinical trials evaluating safety and tolerance of the drug in patients with solid tumors.
The mechanism of caspase inhibition by IAPs is best described for XIAP, which inhibits caspase-3, -7 and -9. XIAP contains three separate BIR domains. BIR2 and the linker to the N-terminal BIR1 domain are responsible for binding active caspase-3 and -7. XIAP potently inhibits these caspases by masking their substrate binding site.85, 86 In contrast, inhibition of caspase-9 solely depends on the BIR3 domain and flanking regions.87, 88 Cleavage of caspase-9 results in the exposition of an IAP-binding motif (IBM) that fits into a hydrophobic pocket of the BIR3 domain. Homologous domains have been discovered in SMAC (second mitochondria-derived activator of caspases), the serine protease Omi/HtrA2 as well as in the Drosophila proteins Reaper, Hid, Grim, Sickle and Jafrac2. SMAC and Omi/HtrA2 are nuclear-encoded proteins residing in mitochondria. The removal of the mitochondrial targeting sequence reveals the IBM comprising four amino acids at the new N-terminus. After induction of the intrinsic apoptotic pathway, both proteins are released into the cytosol. Most interesting with respect to therapeutic intervention is the fact that the IBMs of SMAC and Omi/HtrA2, termed the ‘knob’, also fit into the hydrophobic groove of XIAP. Therefore, SMAC is able to replace and release caspase-9 from the XIAP-inhibitory complex.
Cancer therapeutic approaches employing SMAC peptides or SMAC-mimetic drugs are currently being developed by several companies in order to inhibit IAPs and to restore caspase activity. So far, approaches involving SMAC-like peptides often failed to induce apoptosis as single agents, but nevertheless sensitized cancer cell lines or xenograft mouse models of human glioma and non-small cell lung cancer to TRAIL or chemotherapy.89, 90, 91, 92, 93 Similarly, a nonpeptidic SMAC-mimetic drug (an oxazoline derivative), which was designed by in silico analysis of the SMAC-IBM conformation, could target XIAP, cIAP1 and cIAP2, and synergized with TNF and TRAIL in the killing of cultured tumor cells.94 Sun et al.95 synthesized structure-based SMAC-mimetic compounds, which bound XIAP and enhanced cisplatin-induced apoptosis in human prostate cancer cells, but did not induce apoptosis on their own.
Very recently, capped tripeptides have been developed based on the NMR structure of the Smac/XIAP-BIR3 complex.96 These agents bound to the BIR3 domain of XIAP with high affinity, exhibited cytotoxicity for a range of human tumor cell lines and even slowed down the growth of established breast cancer in a xenograft mouse model. Interestingly, Nikolovska-Coleska et al.97 discovered a low-molecular weight, cell-permeable compound of the Japanese Ardisia herb, termed embelin, through a structure-based computational screen of a herbal medicine database. Embelin activated caspase-9 and induced apoptosis in XIAP-overexpressing prostate cancer cells and had only minor effects on normal epithelial cells. Thus, embelin might serve as a lead compound for the development of SMAC agonists.
SMAC-mimicking small molecules developed by Idun Pharmaceuticals in collaboration with Abbott are already in preclinical testing. However, the treatment of cancer with SMAC agonists still encounters some unresolved questions. For instance, the binding of the SMAC knob in the hydrophobic pocket might be sufficient to derepress caspase-9, but small SMAC peptides often bind to XIAP less efficiently as compared to the wild-type (wt) protein. A second interaction domain of SMAC and BIR3 might play an important role and might be useful if taken into consideration when designing suitable SMAC mimetics.98 The second interaction interface might also explain the finding that an alternatively spliced cytosolic form of SMAC (SMACβ) lacking the knob induces apoptosis to a similar extent as the wt SMAC protein.99 Moreover, IAP-independent functions could account for the antiapoptotic effect of the truncated SMAC proteins.
In addition to caspase-9, targeting of the interaction of XIAP with caspase-3/7 might be even more promising. Schimmer et al.100 employed an elegant enzyme derepression assay to screen for polyphenylurea-based XIAP antagonists. In a fluorigenic substrate assay, a combinatorial library was screened for compounds that could overcome XIAP-mediated repression of caspase-3 activity. The investigators identified several compounds demonstrating a cytotoxic effect predominantly in cancer, but not in nontransformed cells. The observed tumor specificity was suggested to be due to a higher level of already activated but IAP-bound caspases in tumor cells compared to normal cells. Furthermore, the identified polyphenylurea derivatives induced apoptosis in cultured leukemia cells as well as growth suppression of xenografted colon cancer. Even more importantly, overexpression of Bcl-2, Bcl-xL or BI-1 (Bax inhibitor-1) did not alter the sensitivity of cells to the phenylurea compounds, whereas the sensitivity to traditional chemotherapeutic drugs was, as expected, reduced.101
The targeting of IAPs for clinical use is as yet hampered by the limited information on correlations of IAP expression with certain diseases. High expression levels of XIAP have been correlated with poor prognosis in acute myeloid leukemia patients,102 and overexpression of cIAP1 might be associated with some esophageal squamous cell carcinomas.103 More direct genetic evidence comes from a translocation, t(11;18)(q21;q22), found in approximately 50% of Helicobacter pylori-induced mucosa-associated lymphoid tissue (MALT) B-cell lymphomas.104 This translocation causes the expression of a cIAP2-MALT1 fusion protein, with cIAP2 comprising only the two BIR domains and missing the RING and CARD domains. The fusion protein was demonstrated to mediate constitutive nuclear factor-kappaB (NF-κB) stimulation, thereby activating its own transcription in a positive feedback loop. This mechanism presumably renders the tumor independent of the bacteria-caused NF-κB activation and might explain why those tumors no longer respond to antibiotics.105 Therefore, this tumor entity might be a suitable target for IAP-based therapy.
However, drug inhibition of only single IAP interactions might not be sufficient to induce significant in vivo effects, as certain tumors show a simultaneous upregulation of several IAP members.106 In addition, IAP proteins themselves might counteract the therapy: the IAP member ML-IAP, for instance, has only a single BIR domain with a high affinity to SMAC, but with a poor caspase-inhibitory activity. ML-IAP could therefore serve as a sink for applied SMAC agonists, a caveat that might be especially relevant in chemoresistant melanoma, where overexpression of ML-IAP is often observed.107
XIAP, cIAP1, cIAP2, ILP-2 and ML-IAP contain, in addition to one or three BIR domains, also a RING domain associated with E3 ubiquitin ligase activity. RING proteins can mediate the degradation of other proteins and themselves via the proteasome pathway. The RING domain of XIAP has been shown to target XIAP itself, but also caspase-3 for degradation.108 The RING domain might be also involved in caspase-independent antiapoptotic functions of XIAP. Silke et al.109 demonstrated that XIAP mutants, which were no longer able to bind to caspase-3 or -9, still retained antiapoptotic activity. Therefore, BIR2- or BIR3-targeting compounds might not achieve full ablation of XIAP's antiapoptotic effects in cancer cells. Interestingly, another XIAP interacting factor XAF1 is downregulated in various tumor cell lines.110 XAF1 is a negative regulator of XIAP like SMAC, but seems to achieve this effect through the complete removal of XIAP from the cytosol by sequestering it in the nucleus.
Additional complexity is probably yet to surface when interactions with other proteins and post-translational modifications of IAPs are further analyzed. XIAP, for instance, interacts with the type I TGF-β receptor and consequently activates NF-κB and JNK signaling, even though recent studies in XIAP-deficient mice show that this interaction is not required for TGF-β signaling. XIAP also interacts with type I bone morphogenic protein (BMP) receptor and TAB1, a cofactor of TAK1 kinase, while cIAP1 and cIAP2 interfere with death receptors. Nevertheless, although IAP-based therapeutic approaches still encounter several unresolved issues, they have a clear potential for the treatment of cancers. IAP inhibitors will presumably turn out as effective sensitizers for conventional chemo- or radiation therapy.
The Bcl-2 Family
Proteins of the Bcl-2 family are the core components of the intrinsic mitochondrial apoptotic pathway.111Members of this family can be subdivided into pro- and antiapoptotic proteins. Although the precise mechanisms, by which they regulate apoptosis, are still unclear, the stoichiometric balance of pro- and antiapoptotic proteins of this family seems to be crucial. High concentrations of the antiapoptotic proteins Bcl-2, Bcl-xL, A1 or Mcl-1 can inhibit the mitochondrial apoptotic pathway. As tumor radiotherapy as well as most chemotherapeutic agents uses this pathway to kill cancer cells, overexpression of antiapoptotic Bcl-2 family members confers resistance to treatment. Elevated Bcl-2 levels are frequently found in B-cell malignancies and are often caused by the translocation t(14;18)(q32;q21), which brings the bcl-2 gene under the transcriptional control of the highly active immunoglobulin heavy-chain promoter. Bcl-2 overexpression is not limited only to malignancies of B-lymphocytes but is also frequently found in very different types of human cancers of both hematological and nonhematological origin. In these diseases, Bcl-2 overexpression mostly correlates with poor prognosis. Therefore, Bcl-2 proteins emanated early as a very attractive target for both pro- and antiapoptotic therapeutic approaches.
Diverse strategies have been employed to target Bcl-2 and the related proteins Bcl-xL and Mcl-1 in cancer therapy: (1) antisense techniques to downregulate protein levels, (2) application of BH3 domain peptides or (3) synthetic small-molecule drugs, which interfere with complex formation of anti- and proapoptotic Bcl-2 family members, thereby setting previously quenched proapoptotic proteins on the loose.
First approaches to modulate Bcl-2 levels using antisense constructs came from Reed et al.112, 113 A 20-mer oligonucleotide could kill lymphoma and leukemic cells directly, although it was even more effective when combined with standard chemotherapeutic drugs.114, 115 So far, the initially most promising agent being tested in clinical trials targeting Bcl-2 is genasense, a synthetic stabilized 18-mer antisense oligonucleotide to Bcl-2 mRNA developed by Genta. It binds to the first mRNA codons of the Bcl-2 open reading frame and thereby inhibits its expression. In preclinical trials, genasense reduced Bcl-2 protein levels in breast cancer cells to 97% and displayed highly synergistic effects combined with doxorubicin in a xenograft breast cancer model, whereas treatment with genasense alone only delayed tumor growth. Genasense strongly increased also the efficacy of dacarbazine in a xenograft melanoma mouse model. Tumors were completely ablated in 10 of 13 animals, whereas dacarbazine alone elicited no effect.
Unfortunately, the outcomes of the clinical trials with melanoma patients could not fulfill the hopes raised by those preliminary results. Nevertheless, Aventis and Genta filed a new drug application for genasense in combination with dacarbazine in advanced melanoma, which was granted priority review by the FDA this year. However, a major drawback was the decision of the FDA advisory board, which recommended against genasense as a treatment in melanoma. Board members were concerned about the unproven effectiveness of the drug, as it failed to extend the survival time in the latest phase 3 clinical trial comprising 771 patients, even though it did seem to slow down disease progression. Genta withdrew the application, but further clinical studies involving patients with other tumor diseases are about to be evaluated. Thus, genasense might still be a valuable tool in cancer therapy.
Another interesting antisense approach is directed against Bcl-xL, which is also frequently overexpressed in human tumors. The Bcl-x gene encodes several different proteins as a result of alternative splicing. These variants differ in their apoptotic properties: Bcl-xL blocks apoptosis similar to Bcl-2, whereas Bcl-xS induces cell death. Taylor et al.116 used antisense oligonucleotides coupled to 2′-O-(2-methoxy)ethyl (MOE), which does not induce RNase H-like activity, to circumvent the downregulation of all Bcl-x proteins. Instead, by choosing an antisense sequence complementary to a region proximal to the splice donor site, which is essential for the production of Bcl-xL, they triggered the splicing machinery to produce mainly the short-variant protein. Cancer cells treated with these oligonucleotides displayed a decrease in the Bcl-xL : Bcl-xS ratio and were sensitized for chemotherapy-induced apoptosis.
To optimize the impact of an antisense-based therapy, it would be necessary to simultaneously downmodulate at least both Bcl-2 and Bcl-xL, as many human tumors co-overexpress the two proteins. This could be achieved by applying two separate antisense constructs or, as recently shown, by using antisense oligonucleotides against a sequence that is highly homologous in Bcl-2 and Bcl-xL but missing in Bcl-xS mRNA.117 Such bispecific antisense oligonucleotides effectively killed various cancer cells.
All members of the Bcl-2 family contain one to four conserved sequence motifs, called the Bcl-2 homology (BH1–4) domains. Among the proapoptotic members of the Bcl-2 family, two groups of molecules are distinguished: the multidomain proapoptotic Bcl-2 proteins (Bax, Bak, Bok), which contain three BH domains, and the so-called BH3-only proteins (Bad, Bik, Bid, Bim, Blk, Hrk/DP5, Noxa, Puma), which contain only the single BH3 domain.118 Some BH3-only proteins selectively interact with antiapoptotic Bcl-2 family members, whereas others also interact with proapoptotic members. BH3-only proteins have been proposed to be allosteric regulators of the Bcl-2 proteins and serve as sensors and effectors of apoptotic signaling. The current thought is that BH3-only proteins require at least one Bax-type partner to induce cell death. Both Bax and Bak undergo a conformational change in response to apoptotic stimuli, which mediates their assembly into homomultimers with channel-forming properties in the mitochondrial membrane, resulting in cytochrome c release. This conformational change and multimerization of Bak or Bax is inducible by BH3-only proteins and inhibitable by Bcl-2.
Compared to the aforementioned downregulation of Bcl-2 protein levels, the interference with Bcl-2 protein by BH3 domain peptides or small drugs is certainly a more direct and instantaneous approach. The BH3 domain comprises a 9-amino-acid amphipathic α-helix that binds to a hydrophobic pocket of Bcl-2-like antiapoptotic proteins. Therefore, peptides and small molecules mimicking BH3 domain proteins aim at disrupting this complex, thereby sensitizing cells to apoptosis. The disruption of complexes like Bax/Bcl-2 with BH3 peptides and subsequent cytochrome c release could be successfully demonstrated in vitro. However, major challenges of using peptides as anticancer agents are the poor solubility, the susceptibility to proteolytic degradation as well as the limited cell permeability. To enhance cell entry of BH3 domain peptides, several strategies have been employed like (1) coupling of BH3 peptides to protein transduction domains from HIV Tat protein, HSV VP22 or Drosophila Antennapedia protein (ANT), (2) conjugation to fatty acids or (3) delivery by cationic lipids.
Bak-BH3 coupled to ANT, for instance, rapidly entered HeLa cells and induced apoptosis, whereas a point mutant of the BH3 protein abolishing its Bcl-xL binding had no effect.119 ANT-Bak-BH3 even sensitized Bcl-xL-overexpressing cells to CD95-mediated cell death. Other investigators coupled a Bad peptide (residues 140–165) to decanoic acid to enhance cell permeability.120 The peptide induced apoptosis in HL60 tumor cells, but left normal human peripheral lymphocytes relatively unharmed. Furthermore, it slowed down the growth of human myeloid leukemia in SCID mice. Shangary and Johnson121 introduced BH3 peptides into cells by cationic lipid transfer. Jurkat cells overexpressing Bcl-2 or Bcl-xL were efficiently eradicated by this approach, whereas a Bcl-2 BH3 domain peptide had no effect. Bad-BH3 peptides exhibited by far greater killing in Bcl-xL than in Bcl-2 overexpressing cells, and Bax peptides were slightly more effective in Bcl-2-overexpressing cells. This observation does not only confirm the higher affinity of Bad for Bcl-xL that had been found earlier in vitro122 but also points at a possible limitation, because the efficacy of BH3 peptides might depend on the affinity of a certain BH3 domain for a limited set of target proteins.
Promising results came recently from Letai et al.,123 who demonstrated that some BH3 agonists (Bad and Bik peptides) bind to Bcl-2 and induce displacement of any bound proapoptotic protein, whereas other BH3 peptides from Bid and Bim could directly activate Bax and Bak oligomerization. It was also argued that BH3 peptides induce apoptosis by a Bcl-2-independent pathway due to the α-helical structure, which disrupts the mitochondrial membrane on its own and releases proapoptotic factors. Support for this hypothesis came from a study showing apoptosis induction by an α-helical peptide unrelated to BH3 and by a mutant α-helical Bad peptide that was no longer able to bind to Bcl-2.124 Recently, Walensky et al.125 substituted the Bid BH3 domain with non-natural amino acids on the surface opposite to the interacting region by employing a chemical strategy, called hydrocarbon stapling. This approach resulted in stabilized BH3 peptides, termed SAHBs (stabilized α-helix of Bcl-2 domains), with improved pharmacological properties. SAHBs could induce apoptosis in a variety of leukemic cell lines, and reduced the growth of leukemia xenografts in mice without adverse side effects. Thus, even though the exact mechanism of action remains unclear, BH3 peptides and probably more so small drugs mimicking BH3 peptides open up new promising prospects for cancer treatment.
In addition to BH3 peptides, several small-molecule drugs interfering with Bcl-2/Bcl-xL function have been recently identified by screening of chemical libraries. Tetrocarcin A (TC-A), an antibiotic from Actinomyces, was isolated from a library of natural compounds by virtue of its Bcl-2-antagonizing activity. TC-A could sensitize Bcl-2-overexpressing HeLa cells to death receptor- and drug-mediated apoptosis.126 So far, the cellular target of TC-A is unknown. Cell type-specific susceptibility differences have been reported, since T-ALL cells could be directly killed with TC-A as a single agent, whereas other cells were only sensitized for apoptosis induction.127 Thus, TC-A is obviously an interesting candidate for cancer combination therapies.
Antimycin A3, an inhibitor of the mitochondrial electron transport chain, has been demonstrated to bind to the hydrophobic BH3 pocket and to induce apoptosis in Bcl-2- and Bcl-xL-overexpressing cells. This effect was independent of antimycin's effect on cellular respiration, as a structural derivative of antimycin A3 without respiration-inhibitory properties still promoted cell death.128, 129 Screening of a chemical library for compounds able to bind to the BH3 pocket of Bcl-2 proteins also led to the identification of HA14-1. This compound was shown to compete with Bak for the binding to Bcl-2 and to induce apoptosis in HL60 cells.130 Furthermore, it acted synergistically with MEK inhibitors on leukemic cell lines and might therefore be a valuable therapeutic tool.131
Degterev et al.132 developed an HTS assay measuring the displacement of a fluorescent Bak BH3 peptide from Bcl-xL by fluorescence polarization. From a library of preselected 16 320 compounds, they identified two classes of agents, called BH3 inhibitors (BH3Is), that disrupted Bak–Bcl-xL complexes at low micromolar concentrations. BH3Is could disrupt the Bcl-xL complex with Bax and Bad in intact cells. The BH3I-2 compound directly decreased the mitochondrial transmembrane potential prior to cytochrome c efflux, arguing for a more direct influence of BH3Is on mitochondria.133 Although the affinities of BH3Is to their targets are as yet too low to be used as drugs, they are promising lead compounds for the devise of similar high-affinity compounds.
Other researchers employed computational screens based on the NMR structure of Bcl-xL.134 Of 206 876 organic compounds from the NCI 3D database, 35 potential inhibitory molecules were identified and proposed to bind to the BH3 pocket. The most potent compound induced apoptosis in Bcl-2-overexpressing cancer cell lines, but was less effective in cells with low Bcl-2 expression. Chan et al.135 derived chelerythrine, a natural benzophenanthridine alkaloid, in an HTS assay as an inhibitor of Bcl-xL/Bak-BH3 interaction (IC50 1.5 μM) from a library of natural compounds. Chelerythrine induced apoptosis effectively in Bcl-2- or Bcl-xL-overexpressing cells and triggered cytochrome c release from isolated mitochondria. Altogether, these data suggest that many strategies might be employed to attack Bcl-2 proteins in order to manipulate the apoptotic threshold. Nevertheless, given the incomplete understanding of the mechanisms of action of Bcl-2 proteins, Bcl-2 inhibitors and compounds that mimic BH3 domains will have a great potential in anticancer therapy.
Tumor Suppressor p53
P53 is inactivated by mostly point mutation in more than 50% of human tumors. Since it is crucially involved in apoptosis induction after DNA damage, different strategies have been undertaken to target p53. A promising approach is the restoration of normal functions of mutant p53. The radioprotector amifostine, for instance, can restore the transcriptional activity of some p53 mutants in yeast functional assays, provided that they are conformationally flexible.136 Similarly, introduction of small synthetic molecules, which stabilize p53 in a transcriptionally active state by allosteric modulation of its conformation, is thought to target especially tumor cells with a high accumulation of mutant p53.137 Some reports demonstrated that the transcriptional activity of mutant p53 can be rescued by application of synthetic peptides derived from the C-terminus of p53. These peptides interact with the core domain of mutant p53 and restore growth-suppressing functions of p53.138, 139, 140
Another way of stabilizing p53 is by interfering with the binding of its negative regulator Mdm2 either by employing peptides or synthetic drugs.141, 142 Chalcone derivatives have been identified to exert such an activity, but presumably lack sufficient specificity. The latest drug development in this area were so-called ‘nutlins’ discovered by Roche.143 These imidazoline compounds bind to the p53 pocket of Mdm2 and increase p53 activity. Nutlins were shown to inhibit tumor growth in mice to 90% without accompanying harmful side effects.
Upon reintroduction of wt p53 into p53-deficient cancer cells, tumor growth is suppressed, and synergistic effects with conventional chemotherapy have been demonstrated in several studies. Still, the low efficiency of gene therapy vectors limits the success of this therapeutic approach. Hepatotoxicity associated with systemic application of adenoviral vectors is another drawback. Nevertheless, several clinical trials currently evaluate the delivery of wt p53-expressing adenovirus (Ad-p53) by intratumoral injection. INGN2, an Ad-p53 supported by Invitrogen Therapeutics, is currently being tested in a phase 3 trial for the treatment of head and neck cancer. Schering-Plough is investigating the use of the Ad-p53 virus SCH58500 in advanced ovarian cancer. Ad-p53 has also been proposed for elimination of cancer cells ex vivo out of bone marrow from breast cancer patients receiving high-dose chemo- or radiotherapy before autologous bone marrow transplantation. As contamination of bone marrow by cancer cells is a main cause for tumor relapse, purging the transplant with Ad-p53, which specifically targets tumor epithelial and not bone marrow cells, might provide an effective strategy to reduce the risk of tumor relapse.144 A further development using adenoviral strategies is the specific targeting of p53-deficient tumor cells by the adenovirus mutant ONYX-015. This virus is deficient in E1B protein expression, which normally targets and inactivates p53 in order to promote viral replication. Therefore, this virus is thought to replicate only in cells without wt p53 expression, although this selectivity has been recently questioned.145 Currently, the potential therapeutic use of ONYX-015 in combination with cisplatin and 5-FU is under investigation in phase 2/3 clinical trials for patients with recurrent squamous cell cancers.146 Another viral protein inactivating p53 is the E6 protein of human papillomavirus 16 and 18. E6 targets p53 for proteasomal degradation, thereby rendering the phenotype of the cells p53-null.147 Papillomavirus infection is thought to be causative for about 90% of human cervical cancers and 50% of other anogenital cancers. Synthetic peptide aptamers competing intracellularly for E6 binding with p53 are a promising pharmacological approach that has been shown to induce apoptosis in HPV16-positive cancer cells.148
Targeting of heat shock factor (Hsp)-90 by geldanamycin might be a strategy to restore the degradation of mutant p53 by the proteasome. Hsp90 is a protein chaperone that binds to different ‘client proteins’, including a list of cancer relevant targets such as mutant p53, Raf-1 Akt and others. When geldanamycin binds to Hsp90, it disrupts the Hsp90–client protein complex, leading to degradation of the client proteins. Geldanamycin has been reported to deplete mutant p53 in various tumor cell lines.149, 150 As mutant p53 can antagonize other p53 family members, p63 and p73, stabilize MDM2 and gain additional tumor-promoting functions, depletion of mutant p53 might exert a beneficial effect. Kosan Biochemicals has initiated a first phase 2 monotherapy trial in patients with metastatic melanoma using 17-AAG, a 17-allylamino-17-demethoxy derivative of geldanamycin. 17-AAG is also being evaluated in clinical trials for the treatment of multiple other cancer indications.
Chemo- and radiotherapy are limited by the sensitivity of surrounding normal tissues to the applied genotoxic stress. Life-saving high-dose chemo- or radiotherapeutic treatments are associated with side effects like anemia, infections, vomiting and diarrhea. An ingenious approach to tackle this problem was developed by the group of Gudkov. Most cancers harbor mutated inactive p53, while surrounding normal tissues express functional wt p53. Those patients could benefit from a treatment with the small-molecule drug pifithrin-α, a p53 inhibitor. In normal tissues, pifithrin-α would block p53-mediated transcription and apoptosis, relieving the genotoxic stress of the cells. Indeed pifithrin-α completely rescued mice from doses of radiation that usually kill 60% of the mice, and none of the mice developed tumors in 8 months time.151
Conclusions
In the past decade, the identification of the genes and gene products that regulate apoptosis, together with an increased knowledge about their mechanisms of action, has laid the ground stone for the discovery of new drugs targeting apoptosis. The process of apoptosis is controlled at multiple molecular levels, each of which is influenced by different pro- and antiapoptotic proteins. The various decision points of life and death do not only provide an exciting multitude of molecular targets but also offer a breadth of therapeutic options.
Depending on the molecular target, different strategies are being employed. Biologicals including death ligands or agonistic and antagonistic antibodies that inhibit or trigger death receptor signaling have proven efficacy in various animal models. It will be exciting to see whether TRAIL agonists that have now reached clinical trials will be useful in combating cancer or other diseases, either as a single agent or more likely in combination with classical therapeutic regimens. Among all the apoptosis-based drug targets, strategies that target caspases are at the forefront for blocking apoptosis in numerous diseases. Proof-of-concept data have been obtained in several experimental models revealing that blockade of apoptosis by means of caspase inhibitors is a validated therapeutic strategy. However, still a number of key open questions remain. It is unknown whether caspases have physiological roles beyond that of apoptosis, and if so, whether this would bring the risk for toxicity in using caspase inhibitors. Another open issue is whether preservation of cell survival by caspase inhibitors truly results in preservation of cell function. Although small-molecule drugs have been slow to progress to the clinic, there are now two unrelated caspase inhibitors that have shown promising results in early clinical trials.
At present, many compounds developed to modulate apoptosis are only micromolar inhibitors. Small-molecule drugs or peptides like caspase inhibitors have to be optimized to readily penetrate the cells. There is still a great discrepancy between their in vitro and in vivo efficacy, which decisively limits their current use as therapeutics. Nevertheless, many compounds provide compelling evidence and bring the hope that, with sufficient modification by tools of structural biology and combinatorial chemistry, it might be possible to derive sufficiently potent drugs to inhibit apoptosis. This caveat holds also true for endogenous inhibitors of apoptosis. The IAPs are of particular interest as they are the most powerful intrinsic inhibitors of cell death. Several studies have demonstrated that IAP antagonists are promising candidates for anticancer therapies, by either directly killing cancer cells or by augmenting the established chemotherapy. Finally, setting the threshold of apoptosis susceptibility by modulating the Bcl-2/Bax rheostat is another appealing approach for therapy of cancer and other diseases. Nowadays, small molecules or peptidomimetics that mimic BH3 domains represent prototype therapeutics, and the optimal SAR profile of these compounds remains to be improved. Moreover, it is still unresolved whether Bcl-2 antagonists and BH3 mimetics should exert a selective or a broad-spectrum activity against the various members of the Bcl-2 family.
Many new drugs are currently being developed; however, of the hundreds or so molecules in development, most of them will remain in preclinical state. The relatively low rate of clinical entry associated with these molecules is related to the lack of specificity, low efficacy or susceptibility to drug resistance. These issues are being addressed as our understanding of the field evolves, and therefore exploitation of new targets remains a considerable focus of attention. Given that the field of apoptosis research is not more than 15 years old, the fact that first-generation therapeutics are now in human clinical trials is already a tremendous success.
Abbreviations
- BH:
-
Bcl-2 homology domain
- BIR:
-
baculovirus IAP repeat
- CD95L:
-
CD95 ligand
- DISC:
-
death-inducing signaling complex
- fmk:
-
fluoromethylketone
- GVHD:
-
graft-versus-host disease
- HCV:
-
hepatitis C virus
- HIV:
-
human immunodeficiency virus
- HTS:
-
high-throughput screening
- IAP:
-
inhibitor of apoptosis protein
- IBM:
-
IAP-binding motif
- ICE:
-
interleukin-1β-converting enzyme
- MAb:
-
monoclonal antibody
- MS:
-
multiple sclerosis
- NF-κB:
-
nuclear factor-kappaB
- PEA:
-
Pseudomonas exotoxin A
- SAR:
-
structure–activity relationship
- SMAC:
-
second mitochondrial activator of caspases
- TEN:
-
toxic epidermal necrolysis
- TNF:
-
tumor necrosis factor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
References
Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S and Peter ME (1998) Apoptosis signaling by death receptors. Eur. J. Biochem. 254: 439–459
Micheau O and Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190
Daniel PT, Wieder T, Sturm I and Schulze-Osthoff K (2001) The kiss of death: promises and failures of death receptors and ligands in cancer therapy. Leukemia 15: 1022–1032
Roth W, Isenmann S, Naumann U, Kugler S, Bahr M, Dichgans J, Ashkenazi A and Weller M (1999) Locoregional Apo2L/TRAIL eradicates intracranial human malignant glioma xenografts in athymic mice in the absence of neurotoxicity. Biochem. Biophys. Res. Commun. 265: 479–483
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC and Lynch DH (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5: 157–163
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z and Schwall RH (1999) Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 104: 155–162
Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR and Strom SC (2000) Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 6: 564–567
Nitsch R, Bechmann I, Deisz RA, Haas D, Lehmann TN, Wendling U and Zipp F (2000) Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 356: 827–828
Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, Hooley J, Sherwood S, Pai R, Leung S, Khan L, Gliniak B, Bussiere J, Smith CA, Strom SS, Kelley S, Fox JA, Thomas D and Ashkenazi A (2001) Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat. Med. 7: 383–385
Held J and Schulze-Osthoff K (2001) Potential and caveats of TRAIL in cancer therapy. Drug Resist. Update 4: 243–252
Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP and Zhou T (2001) Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat. Med. 7: 954–960
Eggermont AM and ten Hagen TL (2001) Isolated limb perfusion for extremity soft-tissue sarcomas, in-transit metastases, and other unresectable tumors: credits, debits, and future perspectives. Curr. Oncol. Rep. 3: 359–367
Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A and Corti A (2000) Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13). Nat. Biotechnol. 18: 1185–1190
Neurath MF, Fuss I, Pasparakis M, Alexopoulou L, Haralambous S, Meyer zum Buschenfelde KH, Strober W and Kollias G (1997) Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur. J. Immunol. 27: 1743–1750
Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF and Rutgeerts PJ (1997) A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N. Engl. J. Med. 337: 1029–1035
Sandborn WJ, Feagan BG, Radford-Smith G, Kovacs A, Enns R, Innes A and Patel J (2004) CDP571, a humanised monoclonal antibody to tumour necrosis factor alpha, for moderate to severe Crohn's disease: a randomised, double blind, placebo controlled trial. Gut 53: 1485–1493
Sandborn WJ, Hanauer S, Loftus Jr. EV, Tremaine WJ, Kane S, Cohen R, Hanson K, Johnson T, Schmitt D and Jeche R (2004) An open-label study of the human anti-TNF monoclonal antibody adalimumab in subjects with prior loss of response or intolerance to infliximab for Crohn's disease. Am. J. Gastroenterol. 99: 1984–1989
Kam LY and Targan SR (2000) TNF-alpha antagonists for the treatment of Crohn's disease. Expert Opin. Pharmacother. 1: 615–622
Sandborn WJ and Hanauer SB (1999) Antitumor necrosis factor therapy for inflammatory bowel disease: a review of agents, pharmacology, clinical results, and safety. Inflamm. Bowel Dis. 5: 119–133
Li H, Colbourne F, Sun P, Zhao Z, Buchan AM and Iadecola C (2000) Caspase inhibitors reduce neuronal injury after focal but not global cerebral ischemia in rats. Stroke 31: 176–182
Demjen D, Klussmann S, Kleber S, Zuliani C, Stieltjes B, Metzger C, Hirt UA, Walczak H, Falk W, Essig M, Edler L, Krammer PH and Martin-Villalba A (2004) Neutralization of CD95 ligand promotes regeneration and functional recovery after spinal cord injury. Nat. Med. 10: 389–395
Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J and Krammer PH (2001) Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 8: 679–686
Ichikawa H, Ota K and Iwata M (1996) Increased Fas antigen on T cells in multiple sclerosis. J. Neuroimmunol. 71: 125–129
D’Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS and Antel JP (1996) Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J. Exp. Med. 184: 2361–2370
Hattori K, Hirano T, Miyajima H, Yamakawa N, Tateno M, Oshimi K, Kayagaki N, Yagita H and Okumura K (1998) Differential effects of anti-Fas ligand and anti-tumor necrosis factor alpha antibodies on acute graft-versus-host disease pathologies. Blood 91: 4051–4055
Tsukada N, Kobata T, Aizawa Y, Yagita H and Okumura K (1999) Graft-versus-leukemia effect and graft-versus-host disease can be differentiated by cytotoxic mechanisms in a murine model of allogeneic bone marrow transplantation. Blood 93: 2738–2747
Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D, Hunziker T, Saurat JH, Tschopp J and French LE (1998) Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 282: 490–493
Fischer U, Janicke RU and Schulze-Osthoff K (2003) Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 10: 76–100
Fuentes-Prior P and Salvesen GS (2004) The protein structures that shape caspase-activity, specificity, activation, and inhibition. Biochem. J. 384: 201–232
Rodriguez I, Matsuura K, Ody C, Nagata S and Vassalli P (1996) Systemic injection of a tripeptide inhibits the intracellular activation of CPP32-like proteases in vivo and fully protects mice against Fas-mediated fulminant liver destruction and death. J. Exp. Med. 184: 2067–2072
Kunstle G, Leist M, Uhlig S, Revesz L, Feifel R, MacKenzie A and Wendel A (1997) ICE-protease inhibitors block murine liver injury and apoptosis caused by CD95 or by TNF-alpha. Immunol. Lett. 55: 5–10
Ku G, Faust T, Lauffer LL, Livingston DJ and Harding MW (1996) Interleukin-1 beta converting enzyme inhibition blocks progression of type II collagen-induced arthritis in mice. Cytokine 8: 377–386
Miller BE, Krasney PA, Gauvin DM, Holbrook KB, Koonz DJ, Abruzzese RV, Miller RE, Pagani KA, Dolle RE and Ator MA (1995) Inhibition of mature IL-1 beta production in murine macrophages and a murine model of inflammation by WIN 67694, an inhibitor of IL-1 beta converting enzyme. J. Immunol. 154: 1331–1338
Yaoita H, Ogawa K, Maehara K and Maruyama Y (1998) Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 97: 276–281
Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ and Karl IE (1999) Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc. Natl. Acad. Sci. USA 96: 14541–14546
Springer JE, Azbill RD and Knapp PE (1999) Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat. Med. 5: 943–946
Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, Turk D, Vasiljeva O, Stefe I, Vandenabeele P, Bromme D, Puizdar V, Fonovic M, Trstenjak-Prebanda M, Dolenc I, Turk V and Turk B (2003) Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 10: 881–888
Schotte P, Declercq W, Van Huffel S, Vandenabeele P and Beyaert R (1999) Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 442: 117–121
Okamoto Y, Anan H, Nakai E, Morihira K, Yonetoku Y, Kurihara H, Sakashita H, Terai Y, Takeuchi M, Shibanuma T and Isomura Y (1999) Peptide based interleukin-1 beta converting enzyme (ICE) inhibitors: synthesis, structure activity relationships and crystallographic study of the ICE-inhibitor complex. Chem. Pharm. Bull. (Tokyo) 47: 11–21
Lee D, Long SA, Adams JL, Chan G, Vaidya KS, Francis TA, Kikly K, Winkler JD, Sung CM, Debouck C, Richardson S, Levy MA, DeWolf Jr. WE, Keller PM, Tomaszek T, Head MS, Ryan MD, Haltiwanger RC, Liang PH, Janson CA, McDevitt PJ, Johanson K, Concha NO, Chan W, Abdel-Meguid SS, Badger AM, Lark MW, Nadeau DP, Suva LJ, Gowen M and Nuttall ME (2000) Potent and selective nonpeptide inhibitors of caspases 3 and 7 inhibit apoptosis and maintain cell functionality. J. Biol. Chem. 275: 16007–16014
Chapman JG, Magee WP, Stukenbrok HA, Beckius GE, Milici AJ and Tracey WR (2002) A novel nonpeptidic caspase-3/7 inhibitor, (S)-(+)-5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatin reduces myocardial ischemic injury. Eur. J. Pharmacol. 456: 59–68
Scott CW, Sobotka-Briner C, Wilkins DE, Jacobs RT, Folmer JJ, Frazee WJ, Bhat RV, Ghanekar SV and Aharony D (2003) Novel small molecule inhibitors of caspase-3 block cellular and biochemical features of apoptosis. J. Pharmacol. Exp. Ther. 304: 433–440
Nobel CS, Kimland M, Nicholson DW, Orrenius S and Slater AF (1997) Disulfiram is a potent inhibitor of proteases of the caspase family. Chem. Res. Toxicol. 10: 1319–1324
Caserta TM, Smith AN, Gultice AD, Reedy MA and Brown TL (2003) Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 8: 345–352
Han Y, Giroux A, Grimm EL, Aspiotis R, Francoeur S, Bayly CI, McKay DJ, Roy S, Xanthoudakis S, Vaillancourt JP, Rasper DM, Tam J, Tawa P, Thornberry NA, Paterson EP, Garcia-Calvo M, Becker JW, Rotonda J, Nicholson DW and Zamboni RJ (2004) Discovery of novel aspartyl ketone dipeptides as potent and selective caspase-3 inhibitors. Bioorg. Med. Chem. Lett. 14: 805–808
Becker JW, Rotonda J, Soisson SM, Aspiotis R, Bayly C, Francoeur S, Gallant M, Garcia-Calvo M, Giroux A, Grimm E, Han Y, McKay D, Nicholson DW, Peterson E, Renaud J, Roy S, Thornberry N and Zamboni R (2004) Reducing the peptidyl features of caspase-3 inhibitors: a structural analysis. J. Med. Chem. 47: 2466–2474
Isabel E, Black WC, Bayly CI, Grimm EL, Janes MK, McKay DJ, Nicholson DW, Rasper DM, Renaud J, Roy S, Tam J, Thornberry NA, Vaillancourt JP, Xanthoudakis S and Zamboni R (2003) Nicotinyl aspartyl ketones as inhibitors of caspase-3. Bioorg. Med. Chem. Lett. 13: 2137–2140
Han BH, Xu D, Choi J, Han Y, Xanthoudakis S, Roy S, Tam J, Vaillancourt J, Colucci J, Siman R, Giroux A, Robertson GS, Zamboni R, Nicholson DW and Holtzman DM (2002) Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic–ischemic brain injury. J. Biol. Chem. 277: 30128–30136
Toulmond S, Tang K, Bureau Y, Ashdown H, Degen S, O’Donnell R, Tam J, Han Y, Colucci J, Giroux A, Zhu Y, Boucher M, Pikounis B, Xanthoudakis S, Roy S, Rigby M, Zamboni R, Robertson GS, Ng GY, Nicholson DW and Fluckiger JP (2004) Neuroprotective effects of M826, a reversible caspase-3 inhibitor, in the rat malonate model of Huntington's disease. Br. J. Pharmacol. 141: 689–697
Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, Xanthoudakis S, Roy S, Black C, Grimm E, Aspiotis R, Han Y, Nicholson DW and Karl IE (2000) Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat. Immunol. 1: 496–501
Cai SX, Guan L, Jia S, Wang Y, Yang W, Tseng B and Drewe J (2004) Dipeptidyl aspartyl fluoromethylketones as potent caspase inhibitors: SAR of the N-protecting group. Bioorg. Med. Chem. Lett. 14: 5295–5300
Erlanson DA, Lam JW, Wiesmann C, Luong TN, Simmons RL, DeLano WL, Choong IC, Burdett MT, Flanagan WM, Lee D, Gordon EM and O’Brien T (2003) In situ assembly of enzyme inhibitors using extended tethering. Nat. Biotechnol. 21: 308–314
Choong IC, Lew W, Lee D, Pham P, Burdett MT, Lam JW, Wiesmann C, Luong TN, Fahr B, DeLano WL, McDowell RS, Allen DA, Erlanson DA, Gordon EM and O’Brien T (2002) Identification of potent and selective small-molecule inhibitors of caspase-3 through the use of extended tethering and structure-based drug design. J. Med. Chem. 45: 5005–5022
Allen DA, Pham P, Choong IC, Fahr B, Burdett MT, Lew W, DeLano WL, Gordon EM, Lam JW, O’Brien T and Lee D (2003) Identification of potent and novel small-molecule inhibitors of caspase-3. Bioorg. Med. Chem. Lett. 13: 3651–3655
Harter WG, Albrect H, Brady K, Caprathe B, Dunbar J, Gilmore J, Hays S, Kostlan CR, Lunney B and Walker N (2004) The design and synthesis of sulfonamides as caspase-1 inhibitors. Bioorg. Med. Chem. Lett. 14: 809–812
Karanewsky DS, Bai X, Linton SD, Krebs JF, Wu J, Pham B and Tomaselli KJ (1998) Conformationally constrained inhibitors of caspase-1 (interleukin-1 beta converting enzyme) and of the human CED-3 homologue caspase-3 (CPP32, apopain). Bioorg. Med. Chem. Lett. 8: 2757–2762
Linton SD, Karanewsky DS, Ternansky RJ, Wu JC, Pham B, Kodandapani L, Smidt R, Diaz JL, Fritz LC and Tomaselli KJ (2002) Acyl dipeptides as reversible caspase inhibitors. Part 1: initial lead optimization. Bioorg. Med. Chem. Lett. 12: 2969–2971
Linton SD, Karanewsky DS, Ternansky RJ, Chen N, Guo X, Jahangiri KG, Kalish VJ, Meduna SP, Robinson ED, Ullman BR, Wu JC, Pham B, Kodandapani L, Smidt R, Diaz JL, Fritz LC, von Krosigk U, Roggo S, Schmitz A and Tomaselli KJ (2002) Acyl dipeptides as reversible caspase inhibitors. Part 2: further optimization. Bioorg. Med. Chem. Lett. 12: 2973–2975
Linton SD, Aja T, Allegrini PR, Deckwerth TL, Diaz JL, Hengerer B, Herrmann J, Jahangiri KG, Kallen J, Karanewsky DS, Meduna SP, Nalley K, Robinson ED, Roggo S, Rovelli G, Sauter A, Sayers RO, Schmitz A, Smidt R, Ternansky RJ, Tomaselli KJ, Ullman BR, Wiessner C and Wu JC (2004) Oxamyl dipeptide caspase inhibitors developed for the treatment of stroke. Bioorg. Med. Chem. Lett. 14: 2685–2691
Hoglen NC, Chen LS, Fisher CD, Hirakawa BP, Groessl T and Contreras PC (2004) Characterization of IDN-6556 (3-[2-(2-tert-butyl-phenylaminooxalyl)-amino]-propionylamino]-4-oxo-5-(2,3,5,6-tetrafluoro-phenoxy)-pentanoic acid):a liver-targeted caspase inhibitor. J. Pharmacol. Exp. Ther. 309: 634–640
Canbay A, Feldstein A, Baskin-Bey E, Bronk SF and Gores GJ (2004) The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J. Pharmacol. Exp. Ther. 308: 1191–1196
Valentino KL, Gutierrez M, Sanchez R, Winship MJ and Shapiro DA (2003) First clinical trial of a novel caspase inhibitor: anti-apoptotic caspase inhibitor, IDN-6556, improves liver enzymes. Int. J. Clin. Pharmacol. Ther. 41: 441–449
Natori S, Higuchi H, Contreras P and Gores GJ (2003) The caspase inhibitor IDN-6556 prevents caspase activation and apoptosis in sinusoidal endothelial cells during liver preservation injury. Liver Transplant. 9: 278–284
Hoglen NC, Hirakawa BP, Fisher CD, Weeks S, Srinivasan A, Wong AM, Valentino KL, Tomaselli KJ, Bai X, Karanewsky DS and Contreras PC (2001) Characterization of the caspase inhibitor IDN-1965 in a model of apoptosis-associated liver injury. J. Pharmacol. Exp. Ther. 297: 811–818
MacCorkle RA, Freeman KW and Spencer DM (1998) Synthetic activation of caspases: artificial death switches. Proc. Natl. Acad. Sci. USA 95: 3655–3660
Shariat SF, Desai S, Song W, Khan T, Zhao J, Nguyen C, Foster BA, Greenberg N, Spencer DM and Slawin KM (2001) Adenovirus-mediated transfer of inducible caspases: a novel ‘death switch’ gene therapeutic approach to prostate cancer. Cancer Res. 61: 2562–2571
Xie X, Zhao X, Liu Y, Zhang J, Matusik RJ, Slawin KM and Spencer DM (2001) Adenovirus-mediated tissue-targeted expression of a caspase-9-based artificial death switch for the treatment of prostate cancer. Cancer Res. 61: 6795–6804
Komata T, Kondo Y, Kanzawa T, Hirohata S, Koga S, Sumiyoshi H, Srinivasula SM, Barna BP, Germano IM, Takakura M, Inoue M, Alnemri ES, Shay JW, Kyo S and Kondo S (2001) Treatment of malignant glioma cells with the transfer of constitutively active caspase-6 using the human telomerase catalytic subunit (human telomerase reverse transcriptase) gene promoter. Cancer Res. 61: 5796–5802
Jia LT, Zhang LH, Yu CJ, Zhao J, Xu YM, Gui JH, Jin M, Ji ZL, Wen WH, Wang CJ, Chen SY and Yang AG (2003) Specific tumoricidal activity of a secreted proapoptotic protein consisting of HER2 antibody and constitutively active caspase-3. Cancer Res. 63: 3257–3262
Xu YM, Wang LF, Jia LT, Qiu XC, Zhao J, Yu CJ, Zhang R, Zhu F, Wang CJ, Jin BQ, Chen SY and Yang AG (2004) A caspase-6 and anti-human epidermal growth factor receptor-2 (HER2) antibody chimeric molecule suppresses the growth of HER2-overexpressing tumors. J. Immunol. 173: 61–67
Vocero-Akbani AM, Heyden NV, Lissy NA, Ratner L and Dowdy SF (1999) Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat. Med. 5: 29–33
Roy S, Bayly CI, Gareau Y, Houtzager VM, Kargman S, Keen SL, Rowland K, Seiden IM, Thornberry NA and Nicholson DW (2001) Maintenance of caspase-3 proenzyme dormancy by an intrinsic ‘safety catch’ regulatory tripeptide. Proc. Natl. Acad. Sci. USA 98: 6132–6137
Buckley CD, Pilling D, Henriquez NV, Parsonage G, Threlfall K, Scheel-Toellner D, Simmons DL, Akbar AN, Lord JM and Salmon M (1999) RGD peptides induce apoptosis by direct caspase-3 activation. Nature 397: 534–539
Jiang X, Kim HE, Shu H, Zhao Y, Zhang H, Kofron J, Donnelly J, Burns D, Ng SC, Rosenberg S and Wang X (2003) Distinctive roles of PHAP proteins and prothymosin-alpha in a death regulatory pathway. Science 299: 223–226
Nguyen JT and Wells JA (2003) Direct activation of the apoptosis machinery as a mechanism to target cancer cells. Proc. Natl. Acad. Sci. USA 100: 7533–7538
Zhang HZ, Kasibhatla S, Wang Y, Herich J, Guastella J, Tseng B, Drewe J and Cai SX (2004) Discovery, characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay. Bioorg. Med. Chem. 12: 309–317
Salvesen GS and Duckett CS (2002) IAP proteins: blocking the road to death's door. Nat. Rev. Mol. Cell Biol. 3: 401–410
Deveraux QL and Reed JC (1999) IAP family proteins—suppressors of apoptosis. Genes Dev. 13: 239–252
Ambrosini G, Adida C and Altieri DC (1997) A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3: 917–921
Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL and Choo KH (2000) Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol. 10: 1319–1328
Reed JC and Bischoff JR (2000) BIRinging chromosomes through cell division--and survivin’ the experience. Cell 102: 545–548
Holcik M, Yeh C, Korneluk RG and Chow T (2000) Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene 19: 4174–4177
Sasaki H, Sheng Y, Kotsuji F and Tsang BK (2000) Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 60: 5659–5666
Hu Y, Cherton-Horvat G, Dragowska V, Baird S, Korneluk RG, Durkin JP, Mayer LD and LaCasse EC (2003) Antisense oligonucleotides targeting XIAP induce apoptosis and enhance chemotherapeutic activity against human lung cancer cells in vitro and in vivo. Clin. Cancer Res. 9: 2826–2836
Huang Y, Park YC, Rich RL, Segal D, Myszka DG and Wu H (2001) Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 104: 781–790
Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC and Salvesen GS (2001) Structural basis for the inhibition of caspase-3 by XIAP. Cell 104: 791–800
Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS and Reed JC (1999) Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 18: 5242–5251
Sun C, Cai M, Meadows RP, Xu N, Gunasekera AH, Herrmann J, Wu JC and Fesik SW (2000) NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J. Biol. Chem. 275: 33777–33781
Arnt CR, Chiorean MV, Heldebrant MP, Gores GJ and Kaufmann SH (2002) Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J. Biol. Chem. 277: 44236–44243
Fulda S, Wick W, Weller M and Debatin KM (2002) Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 8: 808–815
Guo F, Nimmanapalli R, Paranawithana S, Wittman S, Griffin D, Bali P, O’Bryan E, Fumero C, Wang HG and Bhalla K (2002) Ectopic overexpression of second mitochondria-derived activator of caspases (Smac/DIABLO) or cotreatment with N-terminus of Smac/DIABLO peptide potentiates epothilone B derivative-(BMS 247550) and Apo-2L/TRAIL-induced apoptosis. Blood 99: 3419–3426
Yang L, Mashima T, Sato S, Mochizuki M, Sakamoto H, Yamori T, Oh-Hara T and Tsuruo T (2003) Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide. Cancer Res. 63: 831–837
Tamm I, Trepel M, Cardo-Vila M, Sun Y, Welsh K, Cabezas E, Swatterthwait A, Arap W, Reed JC and Pasqualini R (2003) Peptides targeting caspase inhibitors. J. Biol. Chem. 278: 14401–14405
Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X and Harran PG (2004) A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 305: 1471–1474
Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Tomita Y, Krajewski K, Roller PP and Wang S (2004) Structure-based design, synthesis, and evaluation of conformationally constrained mimetics of the second mitochondria-derived activator of caspase that target the X-linked inhibitor of apoptosis protein/caspase-9 interaction site. J. Med. Chem. 47: 4147–4150
Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR, Tomaselli KJ, Zou H and Fesik SW (2004) Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J. Med. Chem. 47: 4417–4426
Nikolovska-Coleska Z, Xu L, Hu Z, Tomita Y, Li P, Roller PP, Wang R, Fang X, Guo R, Zhang M, Lippman ME, Yang D and Wang S (2004) Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J. Med. Chem. 47: 2430–2440
Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X and Shi Y (2000) Structural basis of IAP recognition by Smac/DIABLO. Nature 408: 1008–1012
Roberts DL, Merrison W, MacFarlane M and Cohen GM (2001) The inhibitor of apoptosis protein-binding domain of Smac is not essential for its proapoptotic activity. J. Cell Biol. 153: 221–228
Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B, Glinsky G, Scudiero D, Sausville E, Salvesen G, Nefzi A, Ostresh JM, Houghten RA and Reed JC (2004) Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 5: 25–35
Wang Z, Cuddy M, Samuel T, Welsh K, Schimmer A, Hanaii F, Houghten R, Pinilla C and Reed JC (2004) Cellular, biochemical, and genetic analysis of mechanism of small-molecule IAP inhibitors. J. Biol. Chem. 279: 48168–48176
Tamm I, Richter S, Oltersdorf D, Creutzig U, Harbott J, Scholz F, Karawajew L, Ludwig WD and Wuchter C (2004) High expression levels of x-linked inhibitor of apoptosis protein and survivin correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clin. Cancer Res. 10: 3737–3744
Imoto I, Yang ZQ, Pimkhaokham A, Tsuda H, Shimada Y, Imamura M, Ohki M and Inazawa J (2001) Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 61: 6629–6634
Baens M, Maes B, Steyls A, Geboes K, Marynen P and De Wolf-Peeters C (2000) The product of the t(11;18), an API2-MLT fusion, marks nearly half of gastric MALT type lymphomas without large cell proliferation. Am. J. Pathol. 156: 1433–1439
Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV and Dixit VM (2000) Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol. Cell 6: 961–967
Krajewska M, Krajewski S, Banares S, Huang X, Turner B, Bubendorf L, Kallioniemi OP, Shabaik A, Vitiello A, Peehl D, Gao GJ and Reed JC (2003) Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin. Cancer Res. 9: 4914–4925
Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS and Dixit VM (2000) ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol. 10: 1359–1366
Yang Y, Fang S, Jensen JP, Weissman AM and Ashwell JD (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288: 874–877
Silke J, Hawkins CJ, Ekert PG, Chew J, Day CL, Pakusch M, Verhagen AM and Vaux DL (2002) The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3- and caspase 9-interacting sites. J. Cell Biol. 157: 115–124
Fong WG, Liston P, Rajcan-Separovic E, St Jean M, Craig C and Korneluk RG (2000) Expression and genetic analysis of XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics 70: 113–122
Cory S, Huang DC and Adams JM (2003) The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22: 8590–8607
Reed JC, Cuddy M, Haldar S, Croce C, Nowell P, Makover D and Bradley K (1990) BCL2-mediated tumorigenicity of a human T-lymphoid cell line: synergy with MYC and inhibition by BCL2 antisense. Proc. Natl. Acad. Sci. USA 87: 3660–3664
Reed JC, Stein C, Subasinghe C, Haldar S, Croce CM, Yum S and Cohen J (1990) Antisense-mediated inhibition of BCL2 protooncogene expression and leukemic cell growth and survival: comparisons of phosphodiester and phosphorothioate oligodeoxynucleotides. Cancer Res. 50: 6565–6570
Campos L, Sabido O, Rouault JP and Guyotat D (1994) Effects of BCL-2 antisense oligodeoxynucleotides on in vitro proliferation and survival of normal marrow progenitors and leukemic cells. Blood 84: 595–600
Keith FJ, Bradbury DA, Zhu YM and Russell NH (1995) Inhibition of bcl-2 with antisense oligonucleotides induces apoptosis and increases the sensitivity of AML blasts to Ara-C. Leukemia 9: 131–138
Taylor JK, Zhang QQ, Wyatt JR and Dean NM (1999) Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat. Biotechnol. 17: 1097–1100
Zangemeister-Wittke U, Leech SH, Olie RA, Simoes-Wust AP, Gautschi O, Luedke GH, Natt F, Haner R, Martin P, Hall J, Nalin CM and Stahel RA (2000) A novel bispecific antisense oligonucleotide inhibiting both bcl-2 and bcl-xL expression efficiently induces apoptosis in tumor cells. Clin. Cancer Res. 6: 2547–2555
Puthalakath H and Strasser A (2002) Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 9: 505–512
Holinger EP, Chittenden T and Lutz RJ (1999) Bak BH3 peptides antagonize Bcl-xL function and induce apoptosis through cytochrome c-independent activation of caspases. J. Biol. Chem. 274: 13298–13304
Wang JL, Zhang ZJ, Choksi S, Shan S, Lu Z, Croce CM, Alnemri ES, Korngold R and Huang Z (2000) Cell permeable Bcl-2 binding peptides: a chemical approach to apoptosis induction in tumor cells. Cancer Res. 60: 1498–1502
Shangary S and Johnson DE (2002) Peptides derived from BH3 domains of Bcl-2 family members: a comparative analysis of inhibition of Bcl-2, Bcl-x(L) and Bax oligomerization, induction of cytochrome c release, and activation of cell death. Biochemistry 41: 9485–9495
Yang E, Zha J, Jockel J, Boise LH, Thompson CB and Korsmeyer SJ (1995) Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80: 285–291
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S and Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192
Schimmer AD, Hedley DW, Chow S, Pham NA, Chakrabartty A, Bouchard D, Mak TW, Trus MR and Minden MD (2001) The BH3 domain of BAD fused to the Antennapedia peptide induces apoptosis via its alpha helical structure and independent of Bcl-2. Cell Death Differ. 8: 725–733
Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL and Korsmeyer SJ (2004) Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305: 1466–1470
Nakashima T, Miura M and Hara M (2000) Tetrocarcin A inhibits mitochondrial functions of Bcl-2 and suppresses its anti-apoptotic activity. Cancer Res. 60: 1229–1235
Tinhofer I, Anether G, Senfter M, Pfaller K, Bernhard D, Hara M and Greil R (2002) Stressful death of T-ALL tumor cells after treatment with the anti-tumor agent Tetrocarcin-A. FASEB J. 16: 1295–1297
Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, Zhang KY and Hockenbery DM (2001) Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat. Cell Biol. 3: 183–191
Kim KM, Giedt CD, Basanez G, O’Neill JW, Hill JJ, Han YH, Tzung SP, Zimmerberg J, Hockenbery DM and Zhang KY (2001) Biophysical characterization of recombinant human Bcl-2 and its interactions with an inhibitory ligand, antimycin A. Biochemistry 40: 4911–4922
Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES and Huang Z (2000) Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. USA 97: 7124–7129
Milella M, Estrov Z, Kornblau SM, Carter BZ, Konopleva M, Tari A, Schober WD, Harris D, Leysath CE, Lopez-Berestein G, Huang Z and Andreeff M (2002) Synergistic induction of apoptosis by simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute myelogenous leukemia. Blood 99: 3461–3464
Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T and Yuan J (2001) Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat. Cell Biol. 3: 173–182
Feng WY, Liu FT, Patwari Y, Agrawal SG, Newland AC and Jia L (2003) BH3-domain mimetic compound BH3I-2′ induces rapid damage to the inner mitochondrial membrane prior to the cytochrome c release from mitochondria. Br. J. Haematol. 121: 332–340
Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, Long YQ, Roller PP, Yang D and Wang S (2001) Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J. Med. Chem. 44: 4313–4324
Chan SL, Lee MC, Tan KO, Yang LK, Lee AS, Flotow H, Fu NY, Butler MS, Soejarto DD, Buss AD and Yu VC (2003) Identification of chelerythrine as an inhibitor of BclXL function. J. Biol. Chem. 278: 20453–20456
Maurici D, Monti P, Campomenosi P, North S, Frebourg T, Fronza G and Hainaut P (2001) Amifostine (WR2721) restores transcriptional activity of specific p53 mutant proteins in a yeast functional assay. Oncogene 20: 3533–3540
Foster BA, Coffey HA, Morin MJ and Rastinejad F (1999) Pharmacological rescue of mutant p53 conformation and function. Science 286: 2507–2510
Selivanova G, Iotsova V, Okan I, Fritsche M, Strom M, Groner B, Grafstrom RC and Wiman KG (1997) Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 3: 632–638
Selivanova G, Ryabchenko L, Jansson E, Iotsova V and Wiman KG (1999) Reactivation of mutant p53 through interaction of a C-terminal peptide with the core domain. Mol. Cell. Biol. 19: 3395–3402
Kim AL, Raffo AJ, Brandt-Rauf PW, Pincus MR, Monaco R, Abarzua P and Fine RL (1999) Conformational and molecular basis for induction of apoptosis by a p53 C-terminal peptide in human cancer cells. J. Biol. Chem. 274: 34924–34931
Chene P, Fuchs J, Bohn J, Garcia-Echeverria C, Furet P and Fabbro D (2000) A small synthetic peptide, which inhibits the p53–hdm2 interaction, stimulates the p53 pathway in tumour cell lines. J. Mol. Biol. 299: 245–253
Stoll R, Renner C, Hansen S, Palme S, Klein C, Belling A, Zeslawski W, Kamionka M, Rehm T, Muhlhahn P, Schumacher R, Hesse F, Kaluza B, Voelter W, Engh RA and Holak TA (2001) Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry 40: 336–344
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N and Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848
Seth P, Brinkmann U, Schwartz GN, Katayose D, Gress R, Pastan I and Cowan K (1996) Adenovirus-mediated gene transfer to human breast tumor cells: an approach for cancer gene therapy and bone marrow purging. Cancer Res. 56: 1346–1351
Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M and zur Hausen H (1998) Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J. Virol. 72: 9470–9478
Khuri FR, Nemunaitis J, Ganly I, Arseneau J, Tannock IF, Romel L, Gore M, Ironside J, MacDougall RH, Heise C, Randlev B, Gillenwater AM, Bruso P, Kaye SB, Hong WK and Kirn DH (2000) A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat. Med. 6: 879–885
Scheffner M, Huibregtse JM, Vierstra RD and Howley PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75: 495–505
Butz K, Denk C, Ullmann A, Scheffner M and Hoppe-Seyler F (2000) Induction of apoptosis in human papillomaviruspositive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proc. Natl. Acad. Sci. USA 97: 6693–6697
Blagosklonny MV, Toretsky J and Neckers L (1995) Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 11: 933–939
An WG, Schnur RC, Neckers L and Blagosklonny MV (1997) Depletion of p185erbB2, Raf-1 and mutant p53 proteins by geldanamycin derivatives correlates with antiproliferative activity. Cancer Chemother. Pharmacol. 40: 60–64
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV and Gudkov AV (1999) A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285: 1733–1737
Tse E and Rabbitts TH (2000) Intracellular antibody-caspase-mediated cell killing: an approach for application in cancer therapy. Proc. Natl. Acad. Sci. USA 97: 12266–12271
Nor JE, Hu Y, Song W, Spencer DM and Nunez G (2002) Ablation of microvessels in vivo upon dimerization of iCaspase-9. Gene Therapy 9: 444–451
Wu TY, Wagner KW, Bursulaya B, Schultz PG and Deveraux QL (2003) Development and characterization of nonpeptidic small molecule inhibitors of the XIAP/caspase-3 interaction. Chem. Biol. 10: 759–767
Bilim V, Kasahara T, Hara N, Takahashi K and Tomita Y (2003) Role of XIAP in the malignant phenotype of transitional cell cancer (TCC) and therapeutic activity of XIAP antisense oligonucleotides against multidrug-resistant TCC in vitro. Int. J. Cancer 103: 29–37
McManus DC, Lefebvre CA, Cherton-Horvat G, St-Jean M, Kandimalla ER, Agrawal S, Morris SJ, Durkin JP and Lacasse EC (2004) Loss of XIAP protein expression by RNAi and antisense approaches sensitizes cancer cells to functionally diverse chemotherapeutics. Oncogene 23: 8105–8117
Mesri M, Wall NR, Li J, Kim RW and Altieri DC (2001) Cancer gene therapy using a survivin mutant adenovirus. J. Clin. Invest. 108: 981–990
Friedler A, Hansson LO, Veprintsev DB, Freund SM, Rippin TM, Nikolova PV, Proctor MR, Rudiger S and Fersht AR (2002) A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA 99: 937–942
Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG and Selivanova G (2002) Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8: 282–288
Garcia-Echeverria C, Chene P, Blommers MJ and Furet P (2000) Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53. J. Med. Chem. 43: 3205–3208
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Fischer, U., Schulze-Osthoff, K. Apoptosis-based therapies and drug targets. Cell Death Differ 12 (Suppl 1), 942–961 (2005). https://doi.org/10.1038/sj.cdd.4401556
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401556
Keywords
This article is cited by
-
An integrated framework for prognosis prediction and drug response modeling in colorectal liver metastasis drug discovery
Journal of Translational Medicine (2024)
-
Role of NLRP3 Inflammasome and Its Inhibitors as Emerging Therapeutic Drug Candidate for Alzheimer’s Disease: a Review of Mechanism of Activation, Regulation, and Inhibition
Inflammation (2023)
-
Valrubicin decreases cell proliferation in gastric cancer through the apoptosis induction
Molecular & Cellular Toxicology (2023)
-
Novel Caspase-1 inhibitor CZL80 improves neurological function in mice after progressive ischemic stroke within a long therapeutic time-window
Acta Pharmacologica Sinica (2022)
-
Cell-to-cell variability in inducible Caspase9-mediated cell death
Cell Death & Disease (2022)
