
Abstract
Vertebrate neuron cell death is both a normal developmental process and the catastrophic outcome of nervous system trauma or degenerative disorders. Although the mechanisms of such death include an evolutionarily conserved core apoptotic pathway that is highly homologous to that first described by Horvitz and co-workers in Caenorhabditis elegans, it appears that many instances of neuron death additionally require the transcription-dependent induction of proapoptotic molecules. One such proapoptotic transcriptional pathway revealed by studies over the past decade revolves about the transcription factor E2F and those molecules that either regulate E2F activity or that are direct or indirect transcriptional targets of E2F. Many of the molecules associated with the E2F apoptotic pathway in postmitotic neurons also participate in the cell cycle in proliferating cells. Observations in human material and in animal and cell culture models show widespread correlation between changes in expression, activity and subcellular localization of E2F-related cell cycle molecules and developmental and catastrophic neuron death. A variety of experimental approaches support a causal role for such changes in the death process and are beginning to indicate how the neuronal E2F pathway activates the core apoptotic machinery. The discovery and elaboration of the neuronal apoptotic E2F pathway provides abundant targets as well as small molecule candidates for potential therapeutic intervention in nervous system trauma and degenerative disease.
Similar content being viewed by others


Developmental and catastrophic vertebrate neuron death and involvement of the core apoptotic pathway
Death of postmitotic neurons can be either a normal or catastrophic event. During vertebrate development roughly half of all neurons generated die,1,2 and this process ultimately appears to play an important and beneficial role in the appropriate matching of pre- and postsynaptic elements. However, outside of development, loss of neurons – as for example due to physical injury, stroke, anticancer treatments or neurodegenerative disease – is a catastrophic event with clear and often debilitating consequences. Understanding the molecular mechanisms that underlie neuron death thus provides both insight about nervous system development and the opportunity for clinical intervention in a number of neurological disorders.
Among the remarkable and fundamental aspects of the groundbreaking work by Horvitz and coworkers on the molecular mechanism of apoptotic cell death is that the same molecular families that govern death in C. elegans also participate in death of vertebrate neurons. Thus, as elaborated in the accompanying review by Putcha and Johnson,3 homologs of ced3 (caspases), ced9 (Bcl2 family), ced4 (APAF1) and Egl-1 (BH3 domain family) represent the core machinery that regulates the distal portion of the neuronal apoptotic pathway.
In addition to the shared evolutionarily conserved core elements that govern apoptotic cell death in C. elegans and vertebrate neurons, there are also important distinctions. In the developing worm, specific individual cells are targeted to die based on genetically preprogrammed expression of the core apoptotic machinery. In contrast, developmental death of vertebrate neurons is a stochastic process involving selection within apparently equivalent populations of cells. This selection is largely based on access/lack of access to extrinsic signals that either promote or prevent neuron death. Examples include apoptosis elicited by the lack of access to neurotrophic factors that would otherwise prevent neuron death,1,2,4 and death evoked by proapoptotic ligands such as proneurotrophins.5 Thus, death occurs by extinguishing survival pathways (such as those involving Akt activation) and/or by permitting activation of pathways that promote death.
A role for transcriptional pathways in vertebrate neuron apoptosis
To promote death, the absence of trophic support or the presence of proapoptotic ligands must be linked to the evolutionarily conserved core death machinery by appropriate intracellular signaling pathways. Research over the last decade or so has provided the important observation that in a wide variety of cases, activation of the core apoptotic machinery in developing vertebrate neurons requires de novo transcription.6,7,8,9,10 This key discovery has in turn inspired the search for those transcription factors that mediate neuronal death and for the signaling pathways by which such factors are activated. To date several major transcription factors have been found that play required roles in propagating apoptotic signals in neurons. These include c-Jun,7,11 the p53 family,12 Forkhead-FOXO13 and E2F. Once such transcription factors are activated in response to apoptotic stimuli, they regulate genes that ultimately interact with and engage the core apoptotic machinery, thereby driving neuron death.
Although the transcriptional component of neuron death was originally considered in the context of normal development, it has become increasingly clear that this mechanism also applies to the catastrophic death associated with neuronal injury and disease. Our aim in this review will be to focus on one particular proximal transcriptional pathway that regulates neuron cell death, namely that involving cell cycle molecules and E2F. Since E2F appears to be at the hub of this pathway, we will refer to it as the ‘E2F pathway’. As will be reviewed here, ample correlative and direct experimental evidence ties the E2F pathway to neuron death in both developmental and catastrophic neuron death. In this review, we will discuss the intracellular events and associated molecules by which apoptotic stimuli engage E2F in the neuronal death process and the mechanisms as well as the consequences of this involvement. We shall also consider how this pathway is linked to activation of the core apoptotic machinery, and how interference with this pathway might be clinically relevant to preventing neuron death associated with injury, stroke and neurodegenerative disorders.
Cell cycle molecules and E2F as regulators of vertebrate neuron apoptosis
Initial findings implicating cell cycle molecules in neuron death
A variety of studies with non-neuronal cells have supported the idea that there is an overlap between the molecules that regulate the cell cycle and that promote apoptotic death. 14 Observations that forcing quiescent cells into the cycle can induce their death seemed to be particularly relevant to that most quiescent of cells, the postmitotic neuron.15 By the early to mid-1990s, several authors raised the notion that cell cycle molecules might participate in neuron death.16,17,18,19,20,21 Material support came from the discoveries that transcripts encoding cyclin D1, a regulator of the cell cycle G1/S transition, are induced in cultured sympathetic neurons targeted to die by withdrawal of NGF,18 and that induction of cyclin D1 protein precedes death of cerebellar granule cells in lurcher and staggerer mutant mice.20 Further evidence arose from the finding that expression of SV40 T-antigen, which sequesters cell-cycle-related pocket protein family members, causes apoptotic death of postmitotic Purkinje cells.16,22 Additional reinforcement came from findings that proliferative inhibition by a ras dominant-negative (d/n) construct or by a variety of small molecule cell cycle inhibitors rescued cultured neuronal cells from apoptotic stimuli.17,23 Agents that block mitosis (when tested in proliferating cells) at or before the G1/S transition were differentially effective, thus highlighting this particular part of the cycle.23
In the interval since these ‘early’ beginnings, much evidence has accrued to implicate cell cycle molecules in neuron death. However, although a substantial and growing number of mammalian cell cycle molecules have been described, only a subset have been linked to neuron death at this time. These include primarily cyclins, cyclin-dependent kinases (cdks), cdk inhibitors, Rb family members, E2Fs and E2F target genes (see Table 1). In the discussion to follow, we shall consider each of these classes of molecules in turn and review the evidence for their involvement in both developmental and catastrophic death of neurons. We shall then present an integrated model for how such molecules regulate neuron survival and death. We shall also discuss how the neuronal apoptotic E2F pathway interfaces with other transcriptional death pathways and the core apoptotic machinery. Finally, we shall briefly consider potential clinical approaches to preventing catastrophic neuron death suggested by present knowledge of the apoptotic E2F pathway.
Cyclins and neuron death
Cyclins are essentially activating regulatory subunits for a series of kinases (cdks) that coordinate the cell cycle.24,25 Each cyclin is generally present only during a particular portion of the cycle and has specific cdk partners. The five major cyclins, their cognate cdks and the cycle phases with which they are associated in viable proliferating cells are as follows: cyclin A (cdk2; S phase and cdk1; G2/M), B (cdk1; G2/M), D (cdk4 and cdk6; G1), E (cdk2; G1/S) and H (cdk7; all phases). The means by which levels of individual cyclins are regulated during different phases of the cycle in proliferating cells, and how such regulation in turn coordinates orderly progression of the cycle has been detailed elsewhere.24,25 What is particularly relevant about viable neurons is that they are nonmitotic and thus out of the cycle in the phase before G1, designated as G0.
Cyclin D1
One of the first indications that a cell is moving into the cycle from G0 is the appearance of G1-associated cyclin D. For this reason, cyclin D is a potentially major player in any cell-cycle-related event in neurons. There are three D-type cyclins, designated D1, D2 and D3. Of these, almost all present literature on neuron death concerns cyclin D1. As noted above, an early finding in the short history of this field was that NGF deprivation induces cyclin D1 transcripts.18 Since then, neuronal levels of cyclin D1 transcripts and/or protein have been reported to be elevated in response to a wide range of apoptotic stimuli in vitro, in vivo and in neurodegenerative diseases. For culture systems, this includes cerebellar granule neurons switched from culture medium containing depolarizing levels of K+ to one containing low levels of K+26,27,28,29 (we shall henceforth refer to this model as ‘repolarization’), cerebellar granule neurons exposed to kainic acid,30 cortical neurons exposed to beta amyloid protein,31 sensory neurons exposed to cisplatin32 and serum-deprived neuroblastoma cells.33 In the repolarization model, the distribution of cyclin D1 also appeared to change from a diffuse cytoplasmic pattern to a nuclear localization.27
In vivo death models in which cyclin D1 protein and/or transcripts are elevated in affected neurons include spinal cord injury;34 cisplatin treatment;35 ischemia–reperfusion injury of the brain,36,37,38 retina,39 or spinal cord;40 lurcher and staggerer mutant mice;20 kainic acid administration38,41,42 and motoneuron death in mice expressing a mutant form of superoxide dismutase (SOD) linked to amyotrophic lateral sclerosis (ALS).43 Finally, cyclin D1 is reported to be aberrantly expressed in Alzheimer patients in those neurons that are susceptible to the disease44 and in motorneurons of patients with sporadic ALS.45
For a number of the neuron death paradigms listed above, cyclin D1 was induced at the transcriptional level. Surprisingly, the mechanisms by which apoptotic stimuli lead to induction of cyclin D1 in neurons are unknown and it will be important to address this issue in future to provide a full understanding of the apoptotic E2F pathway.
In addition to transcriptional induction, nontranscriptional mechanisms may also come into play with respect to elevation of neuronal cyclin D1. Boutellier et al.29 reported that in the cerebellar granule neuron repolarization model, cyclin D1 protein accumulation is due to diminished proteasomal degradation caused by repolarization and inactivation of a Ca++/calmodulin-dependent pathway. At present, it is unclear whether such a mechanism also pertains to apoptotic paradigms that do not involve repolarization.
Although there is much correlative evidence to suggest that upregulation of cyclin D1 is causally involved in neuron death, only limited direct evidence has been generated thus far to test this hypothesis. Ino and Chiba42 reported that continuous infusion of cyclin D1 antisense (but not of sense) oligonucleotides into the lateral ventricles suppressed neuron death evoked by kainic acid. A cyclin D1 antisense construct also protected cultured cortical neurons from death promoted by beta amyloid protein.31 As reviewed below, a growing body of evidence links cdk4 to neuron death, thus implicating (though not directly demonstrating) involvement of cyclin D.
Cyclin E
In addition to cyclin D, cyclin E has been associated with neuron death in both in vitro and in vivo models and in disease. Cyclin E levels are elevated in beta-amyloid-treated cortical neurons31 and in cultured cerebellar granule neurons induced to die by kainic acid treatment46 or by repolarization.27 Elevated cyclin E has also been detected in neurons of Alzheimer disease patients.47 Such findings would be consistent with movement of the affected neurons towards the G1/S boundary. Moreover, cyclin E expression is regulated by a mechanism sensitive to the elevation of cyclin D48 and could well be a consequence of the latter. Other than evidence for involvement of cdk2 (a cyclin E-associated cdk) in certain death paradigms (see below), data directly linking cyclin E to neuron death are presently lacking.
Cyclin B
Cyclin B has been reported to be elevated in cultured sympathetic neurons stimulated to die by exposure to dopamine49,50 and in cultured retinal ganglion cells induced to die by nerve growth factor-promoted activation of the p75 neurotrophin receptor.51 Cyclin D induction was not detected in either model. In addition, cyclin B is elevated in neurons affected by Alzheimer disease44,52,53,54,55 as well as in a number of neurodegenerative disorders including Down's syndrome, progressive supranuclear palsy and Pick's disease52 and in cerebrovascular disease.56 Considering the association of cyclin B with the G2/M phase of the cell cycle, such observations have led to the suggestion that in some cases neurons advance as far as G2/M before death.44,51,52,53,54,55,57,58,59 There is a growing number of reports that apoptotic stimuli can induce neurons, at least in some cases, to enter S phase as indicated by BrdU incorporation, mitotic figures and markers such as the proliferating cell nuclear antigen (PCNA).20,22,31,46,60,61 However, despite the attractive idea that inappropriately expressed cyclin B may play a role in neuron death, there are presently few experimental findings to support this. As reviewed below, the best support at present comes from the cerebellar granule repolarization model in which cdk1, a cyclin B-activated cdk, has been implicated.
The mechanism by which neuronal cyclin B is induced in response to apoptotic stimuli has not been studied. However, in cycling cells, like cyclin E, cyclin B expression is downstream of events triggered by cyclin D1 and E2F (see below) and it thus appears possible that a similar mechanism may pertain to neurons.
Cdks
Cdks are serine–threonine kinases that are activated by cyclins at specific points in the normal cell cycle and that play a required role in regulating orderly movement from one stage to the next.24,25 The anticipated consequence of cyclin induction in neurons subjected to apoptotic stimuli is activation of their cdk partners and, where monitored, this indeed has been the case. Such activation appears to play an important role in a variety of paradigms of both developmental and catastrophic neuron cell death. We will consider here cdks 1,2,3,4 and 6. Although cdk5 is a member of the cdk superfamily and appears to play a role in neuron survival and death,62 there is little evidence that it directly contributes to the cell cycle and hence we will not consider it here.
Cdk4/6
These cyclin D-regulated cdks promote movement of cells from G0 and G1 toward G1/S and would thus be among the first indicators that postmitotic neurons reactivate portions of the cell cycle machinery.24 Cdk4 levels are relatively unchanged in proliferative cells in which the major means of regulation of activity occurs via regulated expression of cyclin D.24 In contrast, levels of cdk4 protein appear to be relatively low in viable neurons. In PC12 cells, which are converted by nerve growth factor (NGF) from proliferating neuroblast-like cells to a postmitotic sympathetic-neuron-like phenotype,63 cdk4 transcripts and protein are dramatically downregulated with neuronal differentiation64 (J Angelastro and LA Greene, unpublished data). Withdrawal of NGF, which promotes apoptotic death in this system, results in a rapid re-expression of cdk4 (J Angelastro and LA Greene, unpublished data). Additional in vitro models of neuronal death in which cdk4 is upregulated include cisplatin-treated sensory neurons,32 beta amyloid-treated PC12 cells and cortical neurons,65 and cerebellar granule neurons subjected to repolarization.28 Furthermore, elevated cdk4 has been reported in neurons in animal models of spinal cord injury,34 kainic acid-induced excitotoxic stress,42 transient spinal cord ischemia,40 stroke37 and ALS.43 Neurons destined to die in Alzheimer disease also show cdk4 protein induction.44,66 Corresponding elevation of cdk6 has not been examined in most cases and was absent in the ALS model.43 The latter finding raises the possibility that cdk6 contrasts with cdk4 in its response to apoptotic stimuli, although this point warrants further examination in additional models. For this reason, we will refer mainly to cdk4 here, but it should be kept in mind that cdk6 may also be involved in neuron death.
In nearly all cases in which both were examined, elevation of cdk4 accompanied elevation of cyclin D1 expression. This would be anticipated to lead to enhanced cdk4/cyclin D-associated kinase activity. This has been directly confirmed in several models including DNA damage of cultured cortical neurons,67 repolarized cultured cerebellar granule neurons27,28,61,68 and in spinal cord of the mouse ALS model.43 Additionally, as discussed below, Rb family members, which are major cellular substrates for activated cdk4, show enhanced phosphorylation in a number of neuronal death models.
The key question raised by such observations is whether activation of cdk4 can play a role in neuronal death. One approach to this issue has been the employment of small molecule cyclin-dependent kinase inhibitors (CDKIs). Of these, one of the most effective has been flavopiridol, a cell permeant inhibitor with selectivity for cdks 1,2 and 4.69 At concentrations at which flavopiridol blocks cdk4 activity in vitro, it rescues cultured neurons from a wide variety of apoptotic stimuli including NGF withdrawal;70 beta amyloid exposure;65 cytosine arabinoside;67,71 camptothecin72 or UV treatment;67 repolarization;27 the electron transport chain inhibitor 3-nitorproprionic acid41 and proteasome inhibitors.73 Flavopiridol also proved to be protective in an animal stroke model.37 Although supportive, the drawback of such studies is that, as with many small molecules, there is the issue of flavopridol's specificity that appears to include at least several additional targets that might also affect neuron survival.74 On the positive side, flavopiridol, which is currently in clinical trials as an anticancer agent,69,75 appears to be remarkably effective in promoting neuron survival, is well tolerated in patients and thus it or a related compound may prove to be clinically useful as a neuroprotective agent.
An alternative strategy to test the role of cdk4 in neuron death has been to interfere with its function or activity. One approach to this has been to overexpress the CDKI proteins p16(Ink4), p21/Waf and p27/Kip1 in neurons. CDKI proteins bind to and interfere with the kinase activity of cyclin–cdk complexes.76,77 Each of these proved to protect cultured neurons from apoptotic stimuli including NGF deprivation,78 the DNA-damaging agents camptothecin, ara-C and UV67 and exposure to proteasome inhibitors.73 In addition, overexpression of p16(Ink4) protected murine neuroblastoma cells from death caused by serum deprivation33. Significantly, p16/Ink4 is selective for activated cdk4/6 complexes, thus indicating the importance of these cdks in the apoptotic mechanism. Another approach has been to overexpress d/n forms of cdks in neurons. Sindbis-virus delivered d/n cdks 4 and 6 protected cultured neurons from NGF deprivation,78 DNA damage,67 beta amyloid65 and proteasome inhibitors.73 D/n forms of cdk3 were not protective in these models nor was d/n cdk2 except in the case of proteasome inhibition.73 siRNAs and antisense constructs are additional potential means to interfere with cdk expression. Transfection of a cdk4 siRNA represses death of cultured neurons evoked by DNA damage (DX Liu, SC Biswas and LA Greene, unpublished data), while infusion of a cdk4 antisense oligonucleotide suppressed in vivo neuron death triggered by the excitotoxin kainic acid.42
To summarize, both correlative and experimental evidence supports a role for cyclin D1 and cdk4 in death of diverse neuron types as triggered by various stimuli. However, in view of the potential lack of specificity of currently employed small molecule inhibitors and of possible unanticipated interactions of CKIs and d/n cdks with unintended targets, additional approaches such as the expanded use of siRNAs and antisense constructs would appear to be useful. It will also be important to further extend such studies to animal models of developmental and catastrophic neuron cell death.
Cdk2
In proliferating cells, cyclin E–cdk2 complexes are activated at the G1/S interface.24,25,77 In addition to the elevation of cyclin E expression cited above, several pieces of evidence implicate cdk2 in at least some paradigms of neuron death. Cyclin E-associated kinase activity was elevated in repolarized cultured cerebellar granule neurons,27 and cdk2 activity was induced in striatal neurons following in vivo mild cerebral ischemia.79 With regard to functional evidence for involvement in death, d/n cdk2 constructs protected cultured neurons from death induced by beta amyloid31 (but see Giovanni et al.65) and proteasome inhibitors.73 In each of the models cited here, there was also evidence for functional involvement of cdk4, suggesting that both cdk4 and cdk2 can contribute to the death mechanism in the same neurons. However, as noted above, d/n cdk4 provided neuronal protection in several death models in which d/n cdk2 was ineffective, indicating that cdk2 activation is not obligatory for neuron death in all cases.
Cdk1
Cdk1 (also referred to as cdc2) appears during the G2/M phases of the cycle in proliferating cells and is activated by cyclin B.24,25,77,80 Cdk1 is a transcriptional target of E2F125 and is repressed by E2F4/p130 complexes.80 As elaborated below, this means that cdk1 may be a downstream target of the events triggered by elevation of cyclin D1 and activation of cdk4.
Like cyclin B, cdk1 is upregulated in affected neurons in Alzheimer disease53 as well as in neurons affected by a number of additional neurodegenerative disorders.52 Immunostaining with a specific antibody for phosphoepitopes further indicates the presence of activated cdk1 in AD neurons.53 Cdk1 is also elevated in experimental neuron death models including Purkinje cells of transgenic animals expressing T-antigen,81 dopaminergic substantia nigral neurons induced to die in vivo by axotomy or by treatment with 6-hydroxydopamine,82 and cerebellar granule cells undergoing in vivo developmental cell death or repolarization-induced death In vitro.83
Aside from a correlative association, experimental evidence for involvement of cdk1 in at least one paradigm of neuron death arises from the observation that transfection of cultured cerebellar granule cells with d/n cdk1 or with a cdk1 antisense oligonucleotide suppressed apoptosis caused by repolarization.83 In this model, a cdk2 antisense was reported to be ineffective in preventing death. Interestingly, examination of a different model of cerebellar neuron death (growth factor withdrawal) indicated no involvement of cdk1.84 It thus will be important in future to test the extent to which cdk1 plays a functional role in additional neuron death paradigms.
The mode by which cdk1 elevation/activation can promote neuron death has also come under consideration. Immunohistochemistry has revealed the presence of shared phosphoepitopes in proliferative M-phase cells and in the neurofibrillary tangles that characterize, and that are thought to contribute to the pathology of, Alzheimer disease.53,59,85 It has thus been suggested that activated cdk1 phosphorylates targets such as tau protein and nucleolin that in turn contribute to neurofibrillary tangles and to neuron degeneration in Alzheimer disease.53,59,85 The study of repolarized cultured cerebellar granule cells revealed a rather different mechanism by which cdk1 contributes to neuron death. In this case, the BH3 domain proapoptotic BAD was a target for cdk1. Phosphorylation of BAD by cdk1 led to its movement to mitochondria where it appeared to participate in the death mechanism by binding to BclXL,83 thereby contributing to activation of the core apoptotic mechanism.
Small molecule cdk inhibitors – evidence but not proof for cdk involvement in neuron death
Olomoucine and roscovitine are purine analog cdk inhibitors that have neuroprotective actions in a variety of in vitro and in vivo neuron death models.27,51,70,79,86,87,88 Although such findings have been interpreted as supporting a role for cdk activity in neuron death, as in the case of flavopiridol, these molecules have less than absolute specificity89 and hence such conclusions, while suggestive, must be viewed with caution and as subject to verification by alternative experimental approaches.
CDKIs: do they have a role in neuron survival and death?
As reviewed above, there are several classes of naturally occurring CDKIs that have been exploited to probe the functional roles of active cdks in neuron death. The protective actions of transfected, exogenous CDKIs suggest that sufficiently high levels of endogenous CDKIs could also serve to protect neurons from death even in the face of cdk activation in response to apoptotic stimuli; conversely loss of endogenous CDKIs might permit elevated cdk activity and neuron death. In this light, it is of interest that levels of the CDKI p27/Kip1 fell substantially in cultured cerebellar granule neurons after repolarization27,28 and in cultured neocortical neurons subjected to oxygen–glucose deprivation.79 In addition, p16/Ink4a is lost from striatal neurons induced to die in vivo by mild cerebral ischemia.79 Of additional relevance, immature CNS neurons of mice null for the CDKIs p19/Ink4d and p27/Kip1 re-entered the cell cycle and underwent mitosis and this was accompanied by elevated levels of neuron death.90 Such findings support the idea that CDKIs play an important role in suppressing cdk activity in postmitotic neurons, and that they function to prevent cell cycle re-entry as well as cdk-dependent apoptosis. The limited number of present observations strongly indicates that further experimental studies are in order to test whether loss of CDKIs is a general response to apoptotic stimuli and is required for neuron death.
An additional intriguing observation regarding CDKIs is that p16/Ink4 levels (as well as those of cdk4) are apparently elevated in neurons of Alzheimer disease patients.66 A possible interpretation of this is that p16 elevation is a compensatory attempt to protect these neurons and that this explains, at least in part, the relatively slow loss of neurons in the disease.
Rb/pocket proteins and neuron death
The apparent widespread involvement of activated cdks 2,4 and perhaps 6 in neuron death raises the question of the identities of their relevant substrates. In proliferating cells, major targets for these G1/S kinases are members of the Rb (also designated pocket protein) family (see Stevaux and Dyson91 for review). The three members of this family are pRb, p107 and p130. Pocket proteins bind to members of the E2F family of transcription factors and act as repressors of E2F-mediated gene expression. There are at least six E2F proteins of which E2F1-5 interact with pocket proteins. There is a degree of specificity in these interactions in that pRb appears to bind E2F1-5, while p107 and p130 preferentially bind E2F4 and E2F5. Pocket proteins inhibit expression of E2F-regulated genes by two mechanisms. One is by binding to and directly blocking the E2F activation domain. The other is by recruiting chromatin-modifying proteins (such as histone deacetylases, methyltransferases and Polycomb group proteins) to E2F-binding sites, thereby causing direct gene repression. The way in which cdks come into this picture is that when they phosphorylate pocket proteins, the latter lose their capacity to bind E2F proteins. The consequences of this are that E2F is liberated to promote gene activation and that repressive E2F–pocket protein complexes are lost, resulting in gene derepression. The roles of activation and derepression of E2F-regulated genes to the cell cycle and to cell death will be discussed in a later section below.
Observations in a variety of systems indicate that cdk activation evoked by apoptotic stimuli does indeed lead to neuronal pocket protein phosphorylation. Stimuli including camptothecin,92 cisplatin,32 beta amyloid,31 repolarization27,68 and proteasome inhibitors73 promote pRb phosphorylation in cultured neurons. Similar findings pertain to p107.92 In the cases tested, such Rb phosphorylation was blocked by the cdk4/6 inhibitor flavopiridol.73,92 Phosphorylation of Rb/p107 was followed by a substantial decline in total Rb/p107 protein and this effect was blocked by caspase inhibitors.68,92 Such observations indicate that activation of cdk4/6 induces Rb/p107 phosphorylation in neurons, and that this in turn permits their caspase-dependent degradation.
Elevated pRb phosphorylation has also been reported to occur in animal models of neuron death including stroke,37 SOD-induced ALS43 and kainic acid-induced excitotoxic stress.41 Similar observations have been made in affected neurons of patients afflicted with Alzheimer disease93 and ALS.45 In the ALS model, phospho-Rb coimmunoprecipitated with cdk4, but not the noncycle-related cdk5,43 while pRb phosphorylation in the stroke model was blocked by flavopiridol.37 Such findings support a role for cdk4 in the observed in vivo phosphorylation of pRb.
Although initial observations did not indicate effects of apoptotic stimuli on the third member of the pocket protein family, p130,92 re-examination in cultured neurons reveals that DNA-damaging agents and NGF deprivation do indeed affect this protein. Cellular subfractionation shows that hypophosphorylated p130 is present in nuclei, whereas the hyperphosphorylated forms of p130 reside in the cytoplasm. In response to apoptotic stimuli, the hypophosphorylated nuclear p130 is rapidly lost while the cytoplasmic forms are little if any changed (DX Liu and LA Greene, unpublished data). This change was blocked by flavopiridol, consistent with a requirement for cdk4/6 activation.
One would anticipate that the functions of pocket proteins would be substantially altered by the effects of apoptotic stimuli on their phosphorylation, levels and subcellular localization. If this is the case, then do such changes play a role in neuron apoptosis? One strong indication of this is the observation that targeting SV40 T-antigen to Purkinje neurons in vivo causes their death.16,22,92 T-antigen binds pocket proteins and disrupts their interactions with endogenous partners such as E2F proteins. In support of such a mechanism, T-antigen that was mutated to inhibit its capacity to bind pocket proteins did not affect Purkinje cell survival.22 If functional loss of pocket proteins promotes neuron death, then their replacement should be protective. To test this, neurons have been transfected with excess pRb or with phosphorylation-resistant pRb mutants. Such constructs protect cultured neurons from diverse stimuli such as DNA-damaging agents, NGF deprivation and proteasome inhibitors.73,92,94
The role of pocket proteins in neuron death and survival raises the question as to which member(s) of the family participate in this action. Most attention has been given to pRb; this protein has been widely studied for its role in the G1/S transition25,77,91 and it is present in postmitotic neurons.95 Although early reports with pRb-null mice suggested a role for pRb in neuron survival, recent studies indicate that loss of this gene causes defects in cell cycle, but does not promote death of postmitotic neurons.96,97 In line with the low levels of p107 in neurons,98,99 targeted deletion of this gene in mice yields no observable effect on neuron survival.100 In addition, recent work indicates that pRb and p107 are not required for survival of postmitotic Purkinje neurons.101 By contrast, p130 is at relatively high levels in neurons98,102 and p130-null mice show massive strain-dependent neuronal death.103 Thus, existing data appear to favor an important role for p130 in survival of postmitotic neurons.
We recently undertook a series of experiments to further identify the specific pocket proteins(s) that regulate survival and death in neurons (DX Liu and LA Greene, unpublished data). Examination of E2F-binding sites in cultured neurons showed a large predominance of complexes with p130 and little presence of pRb or p107. These E2F–p130 complexes were lost in response to apoptotic stimuli as was nuclear p130. To test whether E2F–p130 complexes play a role in neuron survival and death, p130 expression in cultured neurons was downregulated by antisense and siRNA approaches; this resulted in apoptotic death. A functionally impaired p130 that displaces endogenous p130, but cannot promote gene repression also induced neuron death. In contrast, there was no effect of knocking down pRb expression. Finally, transfection of cultured neurons with a p130 mutant resistant to phosphorylation by cdk4 provided strong protection from NGF deprivation and DNA damage. Taken together, such observations support the hypothesis that p130 is the major pocket protein species that regulates neuron survival and death.
E2F at the hub of neuron death: the case for gene derepression
As discussed above, there are two major consequences of cdk-promoted phosphorylation and loss of cellular/nuclear pocket proteins. These are release of pocket proteins from the E2F transactivation domain, thereby permitting E2F-dependent gene induction, and dissolution of E2F–pocket protein repressor complexes, thereby promoting gene derepression. To distinguish between the activation and derepression mechanisms, neurons have been transfected with mutant E2F1 protein that retains the capacity to bind DNA, but lacks the activation domain. When overexpressed, this construct should displace endogenous E2F complexes from DNA, irrespective of their association with pocket proteins. Moreover, on one hand the mutant E2F should block E2F-mediated gene activation and on the other promote derepression of genes that are repressed by E2F–pocket protein complexes. Thus, the mutant should be protective if neuron death depends on E2F-mediated gene activation, but should compromise survival if death is due to E2F-mediated gene derepression. When tested in several different systems of cultured neurons, the mutant construct promoted death, indicating a derepression mechanism.94,104
Additional experimental observations further support a mechanism of neuron death driven at least in part by derepression of E2F-regulated genes. A ‘decoy’ DNA construct consisting of multiple copies of the E2F-binding site also effectively promoted death of neurons.94 This DNA would be expected to compete with endogenous DNA for binding of E2F and E2F–pocket protein complexes and to promote survival in the case of a transcriptional activation mechanism and death in the case of a derepression mechanism. Furthermore, derepression of E2F responsive reporters occurred in neuronal cells not only (as expected) with the E2F activation mutant and binding site decoy, but also in response to apoptotic stimuli.94
If neuron survival requires E2F–pocket protein-mediated gene repression, how does this occur and what happens in response to apoptotic stimuli? Recent findings indicate that pocket proteins bind, in addition to E2F, chromatin-modifying proteins such as histone deacetylases (HDACs) and methyltransferases.91 When E2F–pocket protein complexes bind DNA, the tethered HDACs and methyltransferases modify nearby chromatin-associated proteins such as histones and by this means gene transcription is repressed. Examination of p130 in viable neurons revealed that it binds at least two such chromatin modifiers, HDAC1 and the methyltransferase Suv39H1; moreover, this association is lost in response to apoptotic stimuli (DX Liu and LA Greene, unpublished data). The importance of these pocket protein-bound modifiers in both gene repression and maintenance of neuron survival was indicated by findings that HDAC inhibitors such as trichostatin A cause both derepression of E2F-regulated genes and neuron death.105 Similar results were achieved when neurons were transfected with a d/n form of methyltransferase Suv39H1 (DX Liu and LA Greene, unpublished data). Such observations support a mechanism in which E2F–pocket protein complexes repress proapoptotic genes in viable neurons by serving as anchors for chromatin modifiers and in which apoptotic stimuli lead to loss of such complexes and consequent derepression of proapoptotic genes.
Does E2F-dependent gene activation have a role in neuron death?
There are multiple reports in which overexpression of exogenous E2F1 causes neuron death.84,104,106,107,108,109 Overexpression of E2F1 also accelerated loss of T-antigen-expressing Purkinje cells in transgenic mice.81 Although such findings can and have been viewed as supporting an E2F-dependent gene activation model of neuron death, there are alternative interpretations. For instance, in addition to binding unoccupied E2F1 activation sites (if any such are available), excess free E2F1 could compete with DNA-bound E2F for pocket proteins or could displace DNA-bound E2F–pocket protein complexes. Either of these actions would promote derepression of E2F-responsive genes and, according to our hypothesis, neuron death.
There are additional observations that appear to indicate a role for E2F1-promoted gene activation in neuron death. E2F1 is upregulated in neurons subjected to apoptotic stimuli in vitro46,68,104,105,107,109 and in vivo37 and is elevated in degenerating neurons of patients with Alzheimer disease and with Down's syndrome.93,110 In addition, neurons treated with an E2F1 antisense68,104 as well as neurons cultured from mice null for E2F1107,109,111,112,113 are at least partially protected from diverse apoptotic stimuli. Although such data can be taken to support a mechanism of E2F1-dependent gene activation, here again an alternative interpretation is possible. The E2F1 gene is repressed during G0 and derepressed when cells move into G1/S.25,91 In proliferating cells, such upregulation of E2F1 leads to activation of genes required for S phase.25,91 In neurons, one can imagine that apoptotic stimuli promote, via activation of cdk4 and phosphorylation of p130, derepression of E2F1 transcription and that this in turn leads to accumulation of E2F1 protein which then participates in death by the mechanisms discussed above for overexpression of exogenous E2F1. That is, E2F1 may be an initial target of apoptotic-stimuli-promoted gene derepression in neurons, which then contributes to death by enhancing further gene derepression or by stimulating gene activation.
Although the relative roles that activation and derepression of E2F-responsive genes play in various paradigms of neuron death have yet to be fully worked out, there is at least one system in which activation appears to be of importance. Recent observations established that repolarization of cerebellar granule cells results in transcriptional activation of cdk1, which in turn phosphorylates and functionally activates pro-apoptotic BAD.83,84 A cdk1 d/n was protective indicating that cdk1 contributes to death in this neuron death model.83 Further work identified the cdk1 promoter, which contains an E2F-binding element, and showed that this promoter was sufficient to drive elevated expression of a reporter in repolarized granule neurons.84 Deleting the region containing the E2F-binding element did not raise the basal expression of the reporter construct, whereas mutation of this site suppressed the response to repolarization. Such findings appear to rule out a derepression mechanism for regulation of cdk1 in this model and to favor E2F-mediated gene activation. Given the reported elevation of cdk1 and its activator cyclin B in a number of paradigms as reviewed above, it will be important to determine whether direct activation of the cdk1 gene by E2F plays a wider role in neuron death beyond the repolarization model. Of relevancy, although E2F1-mediated cdk1 expression was required for repolarization-induced death of granule neurons, cdk1 was not elevated in the same neurons induced to die by withdrawal of a growth factor and, in this case, did not appear to play a role in the death mechanism.84
Given such findings, what are the relative roles of E2F-dependent gene derepression and activation in neuron death? The most appealing interpretation at this time is that derepression of E2F-regulated genes plays a major role in the neuron death mechanism; in at least some cases, this leads to elevation of E2F1 levels that contribute to the death process by transcriptional activation of targets such as cdk1 and perhaps, by driving further gene derepression. The extent to which these alternative mechanisms contribute to neuron death may well vary depending on neuron type and apoptotic stimulus.
What are the targets of the neuronal E2F apoptotic pathway?
Irrespective of the relative roles of E2F-mediated gene activation and derepression in neuron death, a key issue is the identities of the proapoptotic proteins that are regulated by these mechanisms. Presently, the verified targets of E2F activation/derepression in neurons are few and this is an area that should merit intensive investigation in the future. As reviewed above, one transcriptional target to emerge is cdk1, which contributes to death by phosphorylating BAD and promoting its translocation from the cytoplasm to mitochondria. Another apparent target is c-Jun. A variety of studies have reported that apoptotic stimuli lead to elevation of neuronal c-Jun levels and that when c-Jun is activated by phosphorylation (by c-Jun N-terminal kinases), it in turns stimulates transcription of genes that contribute to death.7 Supporting c-Jun as a target of the E2F pathway, camptothecin, a DNA-damaging agent that evokes neuronal apoptosis, induced c-Jun in cultured neurons, and this response was blocked by cdk inhibitors such as flavopiridol and olomoucine and substantially suppressed by d/n forms of cdks 2 and 6.114 In contrast, there was no effect of these reagents on c-Jun phosphorylation. These intriguing observations thus establish a potential link between two major transcriptional pathways that regulate neuron apoptosis. In this case, proapoptotic targets of activated c-Jun should also be indirectly dependent on activation of the apoptotic E2F pathway. On a cautionary note, given the possible nonspecific actions of small molecule cdk inhibitors and of d/n cdks, it will be desirable in future to confirm such findings by alternative approaches. In addition, it will be important to establish whether c-Jun induction is downstream of the E2F pathway in other paradigms of neuron death.
B- and c-myb are transcription factors with proapoptotic activity that are also regulated by E2F.115 Myb promoters contain E2F-binding sites, and myb expression is subject to E2F-dependent repression.116 Application of apoptotic stimuli to cultured neurons results in upregulation of B- and c-myb transcripts as well as protein.11,94 A variety of approaches indicate that this is due to relief of E2F-mediated repression94 that is mediated by binding of E2F–p130 to the myb promoter (DX Liu and LA Greene, unpublished). The possibility that B- and/or c-myb regulated in this manner may play a role in neuron death is supported by the observation that overexpression of exogenous B- or c-myb in neurons results in apoptosis.94 Mybs are transcription factors and so the question arises as to what these regulate in turn that promote neuron death. One promising myb target is BIM, a proapoptotic member of the BH3 family that is upregulated in neurons in response to NGF deprivation117,118 and downregulated by NGF when it induces proliferating PC12 cells to become nonmitotic sympathetic-neuron-like cells.119 The BIM promoter contains several myb-binding sites and a reporter construct driven by this promoter (but not one with mutated myb sites) shows elevated expression in response to apoptotic stimuli in neurons (SC Biswas, DX Liu and LA Greene, unpublished data). Moreover, pharmacologic and molecular interference with the E2F pathway totally blocks elevation of neuronal BIM expression in response to apoptotic stimuli (SC Biswas, DX Liu and LA Greene, unpublished data). Since BIM induction in neurons is partially dependent on the c-Jun pathway,117 it may be that blockade of the E2F pathway inhibits BIM expression by a combination of effects on expression of c-Jun and of mybs.
As recounted here, there are many variations on the theme of how the E2F pathway promotes neuron death. In this light, although there has been progress in identifying targets of E2F-dependent gene activation and derepression that play causal roles in neuron death, it seems highly likely that many more remain to be identified. Advances in generating neuron death models and in detecting changes in gene expression should greatly facilitate this important objective.
Putting it all together: a unified model of how the E2F pathway regulates neuron survival and death
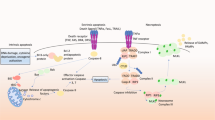
We have reviewed here evidence that proteins otherwise associated with the cell cycle also participate in mediating neuron cell death under a variety of circumstances and that E2F is at the hub of this mechanism. Putting it all together, the following sequential scheme emerges (see Figure 1). In the absence of apoptotic stimuli, viable neurons are in G0 and their levels of cyclin D as well as of cdk4 are low. E2F proteins are complexed by pocket proteins and this results in silencing of E2F-responsive genes, both by inhibition of gene activation and by active gene repression. The latter requires chromatin modifiers that are tethered to the complex. The identities of all the components in the repressor complexes are not known, but include p130, HDAC1 and Suv39H1. The E2F proteins that are involved are unknown at present, but a likely contributor is E2F4, which is a preferred partner for p130 and which is highly expressed in neurons.1,91,120 Such complexes inhibit expression of genes needed for cell cycle entry as well as genes that promote cell death. In response to apoptotic stimuli, levels of cyclin D1 and cdk4 become elevated and as a consequence, neuronal cdk4 activity is greatly enhanced. Activated cdk4 (and perhaps also cdk6) then phosphorylates pocket proteins (of which p130 seems to be the most relevant), which in turn causes dissociation of E2F–pocket protein–chromatin modifier complexes, degradation/cytoplasmic translocation of pocket proteins and loss of gene repression. In what presently appears to be a cell and apoptotic-stimulus-dependent manner, derepressed genes include cyclins E and B, E2F1 and B- and c-myb. Elevated cyclin E activates cdk2 that contributes further to pocket protein phosphorylation and dissolution of E2F–pocket protein complexes. E2F1 activates transcription of cdk1 which, when activated by cyclin B, phosphorylates BAD, thereby permitting the latter to translocate to mitochondria. B- and c-myb induce transcription of BIM. Both phospho-BAD and BIM participate in release of mitochondrial proteins such as cytochrome c, which activate postmitochondrial elements of the core apoptotic pathway. Cyclin B–cdk1 complexes may also phosphorylate cytoskeletal proteins and additional targets that contribute to neuronal degeneration and death. Elevation of c-Jun (which, when activated by phosphorylation, participates in neuron death) also appears to lie downstream of the E2F pathway, but the mechanism of this regulation is unclear. It seems likely that additional proapoptotic downstream targets of the pathway remain to be identified.

Schematic representation of the apoptotic neuronal E2F pathway
A few closing thoughts
On the complexity of neuron death pathways
There are several principles that have emerged from studies on how neurons die. One is that although there are several overarching mechanisms such as involvement of E2F, there is considerable variation in the details of how these play out depending on neuron type and apoptotic stimulus. A good example is the elevation of cdk1 that is associated with repolarization of cerebellar granule neurons, but not with withdrawal of a trophic factor from the same cells. It remains to be seen just how specifics of apoptotic stimulus and neuron type intersect with and drive such variations in the E2F pathway. A second principle is that neuronal apoptotic stimuli often activate diverse death pathways at the same time. Thus, several different transcriptional pathways can be triggered in neurons by apoptotic stimuli, including those dependent on c-Jun, E2F, p53 and FOXO. Moreover, as reviewed here for E2F, even a single such pathway can launch multiple proapoptotic events. One possible reason for such functional redundancy is to insure that death is efficiently achieved. Alternatively, such complexity may be part of a ‘fail-safe’ system that requires at least several independent apoptotic pathways to be launched simultaneously to achieve neuron death. A third and related generalization is that neuron death appears to be a threshold event that can be reached either by very strong activation of a single transcriptional pathway or by the cumulative responses to multiple transcriptional pathways. Experimentally, neuron death can be driven by overexpression of suitable components of any one of the above pathways. Examples for E2F include overexpression of mybs or of the E2F-binding site decoy. However, the observation that blocking any one of the above transcriptional pathways is often sufficient to suppress death promoted by apoptotic stimuli suggests that under physiologic death conditions, no single pathway is sufficiently activated to reach the threshold for death. This may account for widespread expression of elevated E2F pathway molecules observed in neurodegenerative disorders. If such molecules were harbingers of rapidly advancing death, then one might expect to find very few living neurons with cell-cycle-related markers. If on the other hand, additional events, such as activation of p53, must occur to reach an apoptotic threshold, then neurons might persist for relatively long periods of time even though the E2F pathway is activated. A fourth and emerging concept is that multiple transcriptional neuronal apoptotic pathways may converge on single targets. A good case in point is BIM induction. This has been shown to be partially dependent on the JNK pathway117 and recent findings also show regulation by FOXO.121 As mentioned here, BIM additionally appears to be a target of the E2F pathway (SC Biswas, DX Liu and LA Greene, unpublished observations). Such convergence may reflect a mechanism by which multiple transcriptional pathways contribute to reach a threshold level of expression of proapoptotic molecules.
The E2F pathway as a target for therapeutic intervention in catastrophic neuron death
The widespread ectopic expression of cell cycle molecules in susceptible neurons in patients with neurodegenerative disorders and in a variety of disease-related models, coupled with experimental evidence for causative roles of such molecules in neuronal apoptosis, has led many to propose the E2F pathway as a target for therapeutic intervention in catastrophic neuron death.41,52,58,122,123,124,125,126,127 A major challenge is to find means to block the pathway without interfering with normal cell proliferation. One approach is to continue to identify additional pathway components. There is no doubt that such components exist and there are already several intriguing candidates that, due to limited study and length considerations, have been omitted from this review. While the complexity of the E2F pathway and its branch points poses a challenge for those wishing to understand this mechanism in depth, it also provides opportunities by multiplying the number of potential therapeutic targets and thereby, the chances for developing agents that selectively promote neuron survival.
A second approach to therapeutics is to exploit existing agents. It is ironic that most effort toward targeting cell cycle molecules has taken place in the context of neoplasias. It may well be that antiproliferative agents designed to kill rapidly cycling cells will also block neuron death. One example is flavopiridol that is currently in clinical trials for cancer.74,75,128 Although the exact mechanism by which this cdk inhibitor protects neurons is unclear, it does appear to represent a powerful starting point to design drugs that block the E2F pathway and that may repress catastrophic neuron death. Many additional agents have been developed that affect the E2F pathway and it would seem to be of high priority to assess these and derivatives for neuroprotective activity.
Abbreviations
- ALS:
-
amyotrophic lateral sclerosis
- cdk:
-
cyclin-dependent kinase
- CDKI:
-
cyclin-dependent kinase inhibitor
- d/n:
-
dominant-negative
- HDAC:
-
histone deacetylase
- NGF:
-
nerve growth factor
- PCNA:
-
proliferating cell nuclear antigen
- pRb:
-
retinoblastoma protein
- SOD:
-
superoxide dismutase
References
Oppenheim RW (1991) Cell death during development of the nervous system. Annu. Rev. Neurosci. 14: 453–501
Hamburger V (1992) History of the discovery of neuronal death in embryos. J. Neurobiol. 23: 1116–1123
Putcha G and Johnson E (2003) Cell Death Differ. 11: 38–48
Bennet MR, Gibson WG and Lemon G (2002) Neuronal cell death, nerve growth factor and neurotrophic models: 50 years on. Auton. Neurosci. 95: 1–23
Lee R, Kermani P, Teng KK and Hempstead BL (2001) Regulation of cell survival by secreted proneurotrophins. Science 294: 1945–1948
Martin LJ (2001) Neuronal cell death in nervous system development, disease, and injury (review). Int. J. Mol. Med. 7: 455–478
Ham J, Eilers A, Whitfield J, Neame SJ and Shah B (2000) c-Jun and the transcriptional control of neuronal apoptosis. Biochem. Pharmacol. 60: 1015–1021
Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG and Johnson Jr EM (1988) Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 106: 829–844
Oppenheim RW, Prevette D, Tytell M and Homma S (1990) Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: evidence for the role of cell death genes. Dev. Biol. 138: 104–113
Tong L, Toliver-Kinsky T, Taglialatela G, Werrbach-Perez K, Wood T and Perez-Polo JR (1998) Signal transduction in neuronal death. J. Neurochem. 71: 447–459
Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R and Johnson Jr EM (1994) Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 127: 1717–1727
Miller FD, Pozniak CD and Walsh GS (2000) Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 7: 880–888
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868
Vermeulen K, Berneman ZN and Van Bockstaele DR (2003) Cell cycle and apoptosis. Cell Prolif. 36: 165–175
Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ and Hancock DC (1992) Induction of apoptosis in fibroblasts by c-myc protein. Cell 69: 119–128
Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB and Orr HT (1992) Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron 9: 955–966
Ferrari G and Greene LA (1994) Proliferative inhibition by dominant-negative Ras rescues naive and neuronally differentiated PC12 cells from apoptotic death. EMBO J. 13: 5922–5928
Freeman RS, Estus S and Johnson Jr EM (1994) Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron 12: 343–355
Heintz N (1993) Cell death and the cell cycle: a relationship between transformation and neurodegeneration? Trends Biochem. Sci. 18: 157–159
Herrup K and Busser JC (1995) The induction of multiple cell cycle events precedes target-related neuronal death. Development 121: 2385–2395
Rubin LL, Gatchalian CL, Rimon G and Brooks SF (1994) The molecular mechanisms of neuronal apoptosis. Curr. Opin. Neurobiol. 4: 696–702
Feddersen RM, Clark HB, Yunis WS and Orr HT (1995) In vivo viability of postmitotic Purkinje neurons requires pRb family member function. Mol. Cell. Neurosci. 6: 153–167
Farinelli SE and Greene LA (1996) Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J. Neurosci. 16: 1150–1162
Pines J (1995) Cyclins and cyclin-dependent kinases: a biochemical view. Biochem. J. 308 (Part 3): 697–711
Vermeulen K, Van Bockstaele DR and Berneman ZN (2003) The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 36: 131–149
Sakai K, Suzuki K, Tanaka S and Koike T (1999) Up-regulation of cyclin D1 occurs in apoptosis of immature but not mature cerebellar granule neurons in culture. J. Neurosci. Res. 58: 396–406
Padmanabhan J, Park DS, Greene LA and Shelanski ML (1999) Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J. Neurosci. 19: 8747–8756
Martin-Romero FJ, Santiago-Josefat B, Correa-Bordes J, Gutierrez-Merino C and Fernandez-Salguero P (2000) Potassium-induced apoptosis in rat cerebellar granule cells involves cell-cycle blockade at the G1/S transition. J. Mol. Neurosci. 15: 155–165
Boutillier AL, Kienlen-Campard P and Loeffler JP (1999) Depolarization regulates cyclin D1 degradation and neuronal apoptosis: a hypothesis about the role of the ubiquitin/proteasome signalling pathway. Eur. J. Neurosci. 11: 441–448
Giardina SF, Cheung NS, Reid MT and Beart PM (1998) Kainate-induced apoptosis in cultured murine cerebellar granule cells elevates expression of the cell cycle gene cyclin D1. J. Neurochem. 71: 1325–1328
Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F and Sortino MA (1999) Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 13: 2225–2234
Gill JS and Windebank AJ (1998) Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J. Clin. Invest. 101: 2842–2850
Kranenburg O, van der Eb AJ and Zantema A (1996) Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 15: 46–54
Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP and Faden AI (2003) Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann. Neurol. 53: 454–468
Fischer SJ, McDonald ES, Gross L and Windebank AJ (2001) Alterations in cell cycle regulation underlie cisplatin induced apoptosis of dorsal root ganglion neurons in vivo. Neurobiol. Dis. 8: 1027–1035
Guegan C, Levy V, David JP, Ajchenbaum-Cymbalista F and Sola B (1997) c-Jun and cyclin D1 proteins as mediators of neuronal death after a focal ischaemic insult. Neuroreport 8: 1003–1007
Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS, Hakim AM, Ikeda JE and Park DS (2000) Cyclin-dependent kinases as a therapeutic target for stroke. Proc. Natl. Acad. Sci. USA 97: 10254–10259
Timsit S, Rivera S, Ouaghi P, Guischard F, Tremblay E, Ben-Ari Y and Khrestchatisky M (1999) Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy: a modulator of in vivo programmed cell death? Eur. J. Neurosci. 11: 263–278
Kuroiwa S, Katai N, Shibuki H, Kurokawa T, Umihira J, Nikaido T, Kametani K and Yoshimura N (1998) Expression of cell cycle-related genes in dying cells in retinal ischemic injury. Invest. Ophthalmol. Vis. Sci. 39: 610–617
Sakurai M, Hayashi T, Abe K, Itoyama Y, Tabayashi K and Rosenblum WI (2000) Cyclin D1 and Cdk4 protein induction in motor neurons after transient spinal cord ischemia in rabbits. Stroke 31: 200–207
Park DS, Obeidat A, Giovanni A and Greene LA (2000) Cell cycle regulators in neuronal death evoked by excitotoxic stress: implications for neurodegeneration and its treatment. Neurobiol. Aging 21: 771–781
Ino H and Chiba T (2001) Cyclin-dependent kinase 4 and cyclin D1 are required for excitotoxin-induced neuronal cell death in vivo. J. Neurosci. 21: 6086–6094
Nguyen MD, Boudreau M, Kriz J, Couillard-Despres S, Kaplan DR and Julien JP (2003) Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J. Neurosci. 23: 2131–2140
Busser J, Geldmacher DS and Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J. Neurosci. 18: 2801–2807
Ranganathan S and Bowser R (2003) Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am. J. Pathol. 162: 823–835
Verdaguer E, Garcia-Jorda E, Canudas AM, Dominguez E, Jimenez A, Pubill D, Escubedo E, Pallas JC and Camins A (2002) Kainic acid-induced apoptosis in cerebellar granule neurons: an attempt at cell cycle re-entry. Neuroreport 13: 413–416
Nagy Z, Esiri MM, Cato AM and Smith AD (1997) Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl). 94: 6–15
Geng Y, Eaton EN, Picon M, Roberts JM, Lundberg AS, Gifford A, Sardet C and Weinberg RA (1996) Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene 12: 1173–1180
Shirvan A, Ziv I, Machlin T, Zilkha-Falb R, Melamed E and Barzilai A (1997) Two waves of cyclin B and proliferating cell nuclear antigen expression during dopamine-triggered neuronal apoptosis. J. Neurochem. 69: 539–549
Shirvan A, Ziv I, Barzilai A, Djaldeti R, Zilkh-Falb R, Michlin T and Melamed E (1997) Induction of mitosis-related genes during dopamine-triggered apoptosis in sympathetic neurons. J. Neural Transm. Suppl. 50: 67–78
Frade JM (2000) Unscheduled re-entry into the cell cycle induced by NGF precedes cell death in nascent retinal neurones. J. Cell Sci. 113 (Part 7): 1139–1148
Husseman JW, Nochlin D and Vincent I (2000) Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol. Aging 21: 815–828
Vincent I, Jicha G, Rosado M and Dickson DW (1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J. Neurosci. 17: 3588–3598
Vincent I, Bu B, Hudson K, Husseman J, Nochlin D and Jin L (2001) Constitutive Cdc25B tyrosine phosphatase activity in adult brain neurons with M phase-type alterations in Alzheimer's disease. Neuroscience 105: 639–650
Yang Y, Mufson EJ and Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J. Neurosci. 23: 2557–2563
Smith MZ, Nagy Z and Esiri MM (1999) Cell cycle-related protein expression in vascular dementia and Alzheimer's disease. Neurosci. Lett. 271: 45–48
Nagy Z, Esiri MM and Smith AD (1998) The cell division cycle and the pathophysiology of Alzheimer's disease. Neuroscience 87: 731–739
Arendt T, Holzer M, Stobe A, Gartner U, Luth HJ, Bruckner MK and Ueberham U (2000) Activated mitogenic signaling induces a process of dedifferentiation in Alzheimer's disease that eventually results in cell death. Ann. N. Y. Acad. Sci. 920: 249–255
Vincent I, Zheng JH, Dickson DW, Kress Y and Davies P (1998) Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol. Aging. 19: 287–296
Yang Y, Geldmacher DS and Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer's disease. J. Neurosci. 21: 2661–2668
Wu Q, Combs C, Cannady SB, Geldmacher DS and Herrup K (2000) Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol. Aging. 21: 797–806
Smith DS and Tsai LH (2002) Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol. 12: 28–36
Greene LA and Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 73: 2424–2428
Angelastro JM, Klimaschewski L, Tang S, Vitolo OV, Weissman TA, Donlin LT, Shelanski ML and Greene LA (2000) Identification of diverse nerve growth factor-regulated genes by serial analysis of gene expression (SAGE) profiling. Proc. Natl. Acad. Sci. USA 97: 10424–10429
Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R and Park DS (1999) Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F × DP, in B-amyloid-induced neuronal death. J. Biol. Chem. 274: 19011–19016
McShea A, Harris PL, Webster KR, Wahl AF and Smith MA (1997) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am. J. Pathol. 150: 1933–1939
Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM and Greene LA (1998) Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J. Cell Biol. 143: 457–467
Trinh E, Boutillier AL and Loeffler JP (2001) Regulation of the retinoblastoma-dependent Mdm2 and E2F-1 signaling pathways during neuronal apoptosis. Mol. Cell. Neurosci. 17: 342–353
Tan AR and Swain SM (2002) Review of flavopiridol, a cyclin-dependent kinase inhibitor, as breast cancer therapy. Semin. Oncol. 29: 77–85
Park DS, Farinelli SE and Greene LA (1996) Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J. Biol. Chem. 271: 8161–8169
Courtney MJ and Coffey ET (1999) The mechanism of Ara-C-induced apoptosis of differentiating cerebellar granule neurons. Eur. J. Neurosci. 11: 1073–1084
Park DS, Morris EJ, Greene LA and Geller HM (1997) G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J. Neurosci. 17: 1256–1270
Rideout HJ, Wang Q, Park DS and Stefanis L (2003) Cyclin-dependent kinase activity is required for apoptotic death but not inclusion formation in cortical neurons after proteasomal inhibition. J. Neurosci. 23: 1237–1245
Dai Y and Grant S (2003) Cyclin-dependent kinase inhibitors. Curr. Opin. Pharmacol. 3: 362–370
Fischer PM and Gianella-Borradori A (2003) CDK inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs. 12: 955–970
Harper JW (1997) Cyclin dependent kinase inhibitors. Cancer Surv. 29: 91–107
Chellappan SP, Giordano A and Fisher PB (1998) Role of cyclin-dependent kinases and their inhibitors in cellular differentiation and development. Curr. Top. Microbiol. Immunol. 227: 57–103
Park DS, Levine B, Ferrari G and Greene LA (1997) Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J. Neurosci. 17: 8975–8983
Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG and Endres M (2001) Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J. Neurosci. 21: 5045–5053
Taylor WR, Schonthal AH, Galante J and Stark GR (2001) p130/E2F4 binds to and represses the cdc2 promoter in response to p53. J. Biol. Chem. 276: 1998–2006
Athanasiou MC, Yunis W, Coleman N, Ehlenfeldt R, Clark HB, Orr HT and Feddersen RM (1998) The transcription factor E2F-1 in SV40 T antigen-induced cerebellar Purkinje cell degeneration. Mol. Cell. Neurosci. 12: 16–28
El-Khodor BF, Oo TF, Kholodilov N and Burke RE (2003) Ectopic expression of cell cycle markers in models of induced programmed cell death in dopamine neurons of the rat substantia nigra pars compacta. Exp. Neurol. 179: 17–27
Konishi Y, Lehtinen M, Donovan N and Bonni A (2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol. Cell 9: 1005–1016
Konishi Y and Bonni A (2003) The E2F–Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J. Neurosci. 23: 1649–1658
Dranovsky A, Vincent I, Gregori L, Schwarzman A, Colflesh D, Enghild J, Strittmatter W, Davies P and Goldgaber D (2001) Cdc2 phosphorylation of nucleolin demarcates mitotic stages and Alzheimer's disease pathology. Neurobiol. Aging 22: 517–528
Rehen SK, Cid M, Fragel-Madeira L and Linden R (2002) Differential effects of cyclin-dependent kinase blockers upon cell death in the developing retina. Brain Res. 947: 78–83
Bossenmeyer-Pourie C, Chihab R, Schroeder H and Daval JL (1999) Transient hypoxia may lead to neuronal proliferation in the developing mammalian brain: from apoptosis to cell cycle completion. Neuroscience 91: 221–231
Maas Jr JW, Horstmann S, Borasio GD, Anneser JM, Shooter EM and Kahle PJ (1998) Apoptosis of central and peripheral neurons can be prevented with cyclin-dependent kinase/mitogen-activated protein kinase inhibitors. J. Neurochem. 70: 1401–1410
Bain J, McLauchlan H, Elliott M and Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371: 199–204
Zindy F, Cunningham JJ, Sherr CJ, Jogal S, Smeyne RJ and Roussel MF (1999) Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 96: 13462–13467
Stevaux O and Dyson NJ (2002) A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell. Biol. 14: 684–691
Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM and Greene LA (2000) Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J. Neurosci. 20: 3104–3114
Jordan-Sciutto KL, Malaiyandi LM and Bowser R (2002) Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J. Neuropathol. Exp. Neurol. 61: 358–367
Liu DX and Greene LA (2001) Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron 32: 425–438
Ferguson KL and Slack RS (2001) The Rb pathway in neurogenesis. Neuroreport 12: A55–A62
Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK and Slack RS (2002) Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 21: 3337–3346
MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT and Jacks T (2003) Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol. Cell. Biol. 23: 1044–1053
Baldi A, Esposito V, De Luca A, Fu Y, Meoli I, Giordano GG, Caputi M, Baldi F and Giordano A (1997) Differential expression of Rb2/p130 and p107 in normal human tissues and in primary lung cancer. Clin. Cancer Res. 3: 1691–1697
Jiang Z, Zacksenhaus E, Gallie BL and Phillips RA (1997) The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene 14: 1789–1797
LeCouter JE, Kablar B, Hardy WR, Ying C, Megeney LA, May LL and Rudnicki MA (1998) Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol. Cell. Biol. 18: 7455–7465
Marino S, Hoogervoorst D, Brandner S and Berns A (2003) Rb and p107 are required for normal cerebellar development and granule cell survival but not for Purkinje cell persistence. Development 130: 3359–3368
Kusek JC, Greene RM and Pisano MM (2001) Expression of the E2F and retinoblastoma families of proteins during neural differentiation. Brain Res. Bull. 54: 187–198
LeCouter JE, Kablar B, Whyte PF, Ying C and Rudnicki MA (1998) Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development 125: 4669–4679
Hou ST, Cowan E, Walker T, Ohan N, Dove M, Rasqinha I and MacManus JP (2001) The transcription factor E2F1 promotes dopamine-evoked neuronal apoptosis by a mechanism independent of transcriptional activation. J. Neurochem. 78: 287–297
Boutillier AL, Trinh E and Loeffler JP (2003) Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J. Neurochem. 84: 814–828
Azuma-Hara M, Taniura H, Uetsuki T, Niinobe M and Yoshikawa K (1999) Regulation and deregulation of E2F1 in postmitotic neurons differentiated from embryonal carcinoma P19 cells. Exp. Cell Res. 251: 442–451
Hou ST, Cowan E, Dostanic S, Rasquinha I, Comas T, Morley P and MacManus JP (2001) Increased expression of the transcription factor E2F1 during dopamine-evoked, caspase-3-mediated apoptosis in rat cortical neurons. Neurosci. Lett. 306: 153–156
Kobayashi M, Taniura H and Yoshikawa K (2002) Ectopic expression of necdin induces differentiation of mouse neuroblastoma cells. J. Biol. Chem. 277: 42128–42135
O'Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS and Park DS (2000) Induction and modulation of cerebellar granule neuron death by E2F-1. J. Biol. Chem. 275: 25358–25364
Motonaga K, Itoh M, Hirayama A, Hirano S, Becker LE, Goto Y and Takashima S (2001) Up-regulation of E2F-1 in Down's syndrome brain exhibiting neuropathological features of Alzheimer-type dementia. Brain Res. 905: 250–253
Gendron TF, Mealing GA, Paris J, Lou A, Edwards A, Hou ST, MacManus JP, Hakim AM and Morley P (2001) Attenuation of neurotoxicity in cortical cultures and hippocampal slices from E2F1 knockout mice. J. Neurochem. 78: 316–324
Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, Robertson GS, Slack RS and Park DS (2000) E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J. Biol. Chem. 275: 11553–11560
Hou ST, Callaghan D, Fournier MC, Hill I, Kang L, Massie B, Morley P, Murray C, Rasquinha I, Slack R and MacManus JP (2000) The transcription factor E2F1 modulates apoptosis of neurons. J. Neurochem. 75: 91–100
Ghahremani MH, Keramaris E, Shree T, Xia Z, Davis RJ, Flavell R, Slack RS and Park DS (2002) Interaction of the c-Jun/JNK pathway and cyclin-dependent kinases in death of embryonic cortical neurons evoked by DNA damage. J. Biol. Chem. 277: 35586–35596
Oh IH and Reddy EP (1999) The myb gene family in cell growth, differentiation and apoptosis. Oncogene 18: 3017–3033
Catchpole S, Tavner F, Le Cam L, Sardet C and Watson RJ (2002) A B-myb promoter corepressor site facilitates in vivo occupation of the adjacent E2F site by p107 × E2F and p130 × E2F complexes. J. Biol. Chem. 277: 39015–39024
Whitfield J, Neame SJ, Paquet L, Bernard O and Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29: 629–643
Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A and Johnson EM (2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29: 615–628
Biswas SC and Greene LA (2002) Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J. Biol. Chem. 277: 49511–49516
Persengiev SP, Kondova II and Kilpatrick DL (1999) E2F4 actively promotes the initiation and maintenance of nerve growth factor-induced cell differentiation. Mol. Cell. Biol. 19: 6048–6056
Gilley J, Coffer PJ and Ham J (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162: 613–622
Herrup K and Arendt T (2002) Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer's disease. J. Alzheimers Dis. 4: 243–247
Copani A, Sortino MA, Nicoletti F and Giuffrida SA (2002) Alzheimer's disease research enters a ‘new cycle’: how significant? Neurochem. Res. 27: 173–176
Liu DX and Greene LA (2001) Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell Tissue Res. 305: 217–228
McShea A, Wahl AF and Smith MA (1999) Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med. Hypotheses 52: 525–527
O'Hare M, Wang F and Park DS (2002) Cyclin-dependent kinases as potential targets to improve stroke outcome. Pharmacol Ther. 93: 135–143
Raina AK, Zhu X, Rottkamp CA, Monteiro M, Takeda A and Smith MA (2000) Cyclin' toward dementia: cell cycle abnormalities and abortive oncogenesis in Alzheimer disease. J. Neurosci. Res. 61: 128–133
Huwe A, Mazitschek R and Giannis A (2003) Small molecules as inhibitors of cyclin-dependent kinases. Angew. Chem. Int. Ed. Eng. 42: 2122–2138
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Dr G Melino
Rights and permissions
About this article
Cite this article
Greene, L., Biswas, S. & Liu, D. Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ 11, 49–60 (2004). https://doi.org/10.1038/sj.cdd.4401341
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401341
