
Abstract
The goal of the current study was to determine the roles of ATP content, endoplasmic reticulum (ER) Ca2+ stores, cytosolic free Ca2+ (Ca2+f) and calpain activity in the signaling of rabbit renal proximal tubular (RPT) cell death (oncosis). Increasing concentrations (0.3–10 μM) of the mitochondrial inhibitor antimycin A produced rapid ATP depletion that correlated to a rapid and sustained increase in Ca2+f, but not phospholipase C activation. The ER Ca2+-ATPase inhibitors thapsigargin (5 μM) or cyclopiazonic acid (100 μM) alone produced similar but transient increases in Ca2+f. Pretreatment with thapsigargin prevented antimycin A-induced increases in Ca2+f and antimycin A pretreatment prevented thapsigargin-induced increases in Ca2+f. Calpain activity increased in conjunction with ER Ca2+ release. Pretreatment, but not post-treatment, with thapsigargin or cyclopiazonic acid prevented antimycin A-induced cell death. These data demonstrate that extensive ATP depletion signals oncosis through ER Ca2+ release, a sustained increase in Ca2+f and calpain activation. Depletion of ER Ca2+ stores prior to toxicant exposure prevents increases in Ca2+f and oncosis.
Similar content being viewed by others

Introduction
An initial effect of ischemia, hypoxia, or mitochondrial inhibition is the prevention of ATP production. When ATP production falls, protein kinases are unable to phosphorylate phosphoproteins and ATP-dependent ion transporters are inhibited. Further, the loss of oxidative phosphorylation and ATP depletion results in either oncotic (necrotic, rapid) or apoptotic (delayed) cell death, depending on the level of glycolytic ATP production. For example, in cultured mouse renal proximal tubules (RPT), Lieberthal et al.1 determined that an 85% reduction in ATP levels was required to switch the mechanism of cell death from apoptosis to oncosis. RPT have low glycolytic activity and are very susceptible to ischemia- and toxicant-induced oncosis.
Many studies suggest that increases in cytosolic free Ca2+ (Ca2+f) mediate oncosis following anoxia/hypoxia and chemical exposure. For example, studies using concurrent measurement of Ca2+f and cell viability revealed that increases in Ca2+f occur prior to plasma membrane damage.2,3 In addition, decreasing the extracellular Ca2+ concentration reduced the release of lactate dehydrogenase (LDH), a marker of cell death and membrane damage, from rabbit RPT subjected to anoxia or mitochondrial inhibition and rat RPT subjected to hypoxia.4,5,6 Further, chelation of intracellular Ca2+ prevented extracellular Ca2+ uptake and cell death in mitochondrial inhibitor-exposed RPT.7 Finally, evidence supporting a role for Ca2+ in cell injury comes from the observed cytoprotection of Ca2+ channel blockers in renal cell models subjected to anoxia or hypoxia.6,7,8 These studies support an important role for Ca2+ in cell death and suggest that a rise in Ca2+f occurs before extracellular Ca2+ influx.
The endoplasmic reticulum (ER) contains the largest intracellular store of Ca2+ and is replenished by a high-affinity, low-capacity Ca2+-ATPase uptake system (SERCA).9,10 Physiological release of ER Ca2+ results from phospholipase C-mediated inositol trisphosphate (IP3) formation and its subsequent binding to IP3 receptors on the ER.10 In many cells, depletion of ER Ca2+ stores triggers extracellular Ca2+ entry, resulting in a rise in Ca2+F and the refilling of the ER Ca2+ store. This influx pathway is termed capacitative Ca2+ entry or store-operated Ca2+ entry.11 Waters et al.12 suggested that ER Ca2+ release played a role in RPT cell death by demonstrating that depletion of ER Ca2+ stores prior to hypoxia or mitochondrial inhibitor exposure prevented cell death.
Supraphysiological and/or prolonged increases in Ca2+f are thought to activate degradative enzymes such as calpains, Ca2+-activated cysteine proteases. Ischemic/hypoxic injury in brain, kidney, liver and myocardium is thought to be mediated by calpains.13,14,15,16,17,18,19 Using the RPT model, we demonstrated that dissimilar calpain inhibitors decrease RPT cell death produced by a variety of toxicants.7,20,21 However, there is limited data demonstrating increased calpain activity during cell injury and death. Edelstein et al.16 reported an increase in calpain activity in rat RPT subjected to anoxia and Bronk and Gores13 reported an increase in calpain-like protease activity in rat hepatocytes subjected to anoxia. However, increases in calpain activity have not been examined in relation to increases in Ca2+f during cell injury and death. The pathway in Figure 1 illustrates our current hypothesis for the role of ATP content, ER Ca2+ stores, cytosolic free Ca2+ and calpains in cell death. However, several critical questions remain. Is ER Ca2+ released during cell injury and death? What is the signal that results in the release of ER Ca2+? Is calpain activity increased as a result of the ER Ca2+ release? The goal of the studies in this manuscript is to answer these questions.

Hypothesis of Ca2+f increases and calpain activation in RPT during antimycin A treatment. ER Ca2+ release may occur by either phospholipase C (PLC)-generated inositol trisphosphate (IP3) binding to the IP3 receptor on the ER or by inhibition of the ER Ca2+-ATPase by ATP depletion. The early events upon antimycin A exposure include increases in Ca2+f levels due to ER Ca2+ release and extracellular Ca2+ influx and resulting increase in calpain activity. Calpain activation leads to substrate hydrolysis, late stage Ca2+ influx and cell death
Results
Characterization of ER Ca2+ release with ER Ca2+-ATPase inhibitors
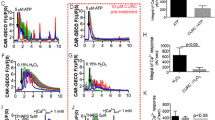
To determine the kinetics of ER Ca2+ release with smooth endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitors, thapsigargin (5 μM) or cyclopiazonic acid (100 μM) was added to RPT suspensions and Ca2+f monitored. Thapsigargin is an irreversible inhibitor and cyclopiazonic acid is a reversible inhibitor of SERCAs.22,23 Inhibition of SERCAs quickly results in increases in Ca2+f through the ER Ca2+ leak. Increases in Ca2+f occurred within 10 s after exposure to thapsigargin or cyclopiazonic acid, resulting in similar peak Ca2+f levels (184±7 nM baseline to 275±20 and 292±13 nM, respectively) before returning to the baseline level (30 s after addition) (Figure 2A,B). Both agents produced similar rises and decays in Ca2+f (Figure 2A,B). To test whether the ER Ca2+ store was completely depleted by 5 μM thapsigargin or 100 μM cyclopiazonic acid, two experiments were performed. In the first, the agents were added again 1 min following the first exposure to thapsigargin. The second addition of thapsigargin or cyclopiazonic acid did not result in a second peak, indicating ER Ca2+ depletion (data not shown). The addition of ionomycin in the absence of extracellular Ca2+ non-specifically releases internal Ca2+ stores. In the second experiment, pretreatment with thapsigargin deceased Ca2+f increases due to ionomycin (10 μM) by 89%. To determine whether extracellular Ca2+ influx contributed to the rise or decay of Ca2+f, extracellular Ca2+ was chelated with EGTA. Decreasing extracellular Ca2+ levels to 100 μM immediately prior to thapsigargin or cyclopiazonic acid did not significantly alter the peak or decay of Ca2+f, suggesting little or no extracellular Ca2+ influx during or immediately after ER Ca2+ depletion with these agents (Figure 2).

The effect of thapsigargin (Thaps) (A) and cyclopiazonic acid (CPA) (B) on cytosolic Ca2+f levels in RPT. Thaps (5 μM) or CPA (100 μM) were added with or without addition of EGTA (900 μM) and readings obtained for an additional 2 min. Shown are representative traces (n=4). Closed circles are without EGTA, open circles are with EGTA
Effect of antimycin A exposure on Ca2+f levels
Antimycin A blocks the electron transport chain and has been used as a model of anoxia (i.e. chemical anoxia).24 Antimycin A caused a concentration-dependent (0.3–10 μM) increase in Ca2+f levels, resulting in a sustained peak by 90 s (Figure 3A,B). Ca2+f measurements taken 15 and 30 min after addition of 10 μM antimycin A revealed that the initial increases in Ca2+f were sustained (data not shown).

The effect of antimycin A (AA) on cytosolic Ca2+f levels in RPT. (A) Antimycin A was added at 15 s and readings obtained for an additional 3 min (a=dimethylsulfoxide control; b=0.3 μM; c=1.0 μM; d=3 μM; e=10 μM AA). Shown are representative traces (n=4). (B) Bars represent the means±s.e. increase above baseline between 60 and 90 s after addition of AA (n=4). Bars with different letters are significantly different from one another (P<0.05)
Effect of antimycin A exposure on ATP levels
Experiments were conducted to determine the concentration- and time-dependent effects of antimycin A on ATP levels in RPT. Within 60 s of addition, antimycin A decreased ATP levels by 90% or greater at all concentrations used (0.3–10 μM) (Figure 4A). Time course experiments revealed that 10 μM antimycin A decreased ATP levels 89% within 30 s and 97% within 2 min (Figure 4B). The antimycin A-induced ATP depletion and increase in Ca2+f levels both occurred within 60 s following antimycin A addition, suggesting that depletion of ATP, the resulting inhibition of the ER Ca2+-ATPase and ER Ca2+ release may account for the increases in Ca2+f.

The effect of antimycin A (AA) on intracellular ATP content. (A) ATP content was determined 1 min after addition of the indicated concentration of AA. (B) ATP content was determined at the indicated time point after exposure to 10 μM AA. Bars represent means±s.e. (n=3). Bars with different letters are significantly different from one another (P<0.05)
Effect of ER Ca2+ depletion on antimycin A-induced Ca2+f increases
To determine whether antimycin A released the thapsigargin-sensitive ER Ca2+ pool and whether this release could account for the Ca2+f increase observed after antimycin A addition in the presence of extracellular Ca2+, thapsigargin was added 100 s after antimycin A and Ca2+f levels were monitored. Thapsigargin addition did not result in additional Ca2+f increases after addition of antimycin A (Figure 5A). Likewise, when antimycin A was added 100 s after the depletion of ER Ca2+ with thapsigargin, antimycin A did not result in an increase in Ca2+f (Figure 5B). To determine whether the early release of ER Ca2+ prevented antimycin A-induced Ca2+f increases over an extended period of time, Ca2+f levels were measured 30 min after antimycin A with and without a 5-min pretreatment with thapsigargin or cyclopiazonic acid. Thapsigargin and cyclopiazonic acid also prevented the antimycin A-induced increases in Ca2+f observed at 30 min (data not shown).

The effect of thapsigargin (Thaps) on antimycin A (AA)-induced increases in Ca2+f. (A) AA (10 μM) was added 100 s prior to Thaps (5 μM). (B) Thaps was added 100 s prior to AA. Shown are representative traces (n=4)
Effect of U73122 on antimycin A-induced Ca2+f increases
The phospholipase C (PLC)-inositol phosphate pathway is a major Ca2+ signaling mechanism in cells.25 Activation of PLC triggers the production of IP3, the binding of IP3 to IP3 receptors on the ER and the release of ER Ca2+. U73122 has been used in a number of cell types to inhibit PLC.26,27,28 Inhibition of PLC results in the blockade of IP3 production and thus serves as a useful pharmacological tool for determining whether IP3-sensitive ER Ca2+ release plays a role in a cell response. To determine whether the antimycin A-induced Ca2+ response is mediated by PLC/IP3, we pretreated RPT with 2 μM U73122 10 min prior to 10 μM antimycin A addition. Pretreatment with U73122 did not alter the antimycin A-induced increase in Ca2+f (data not shown). As a positive control, ATP (50 μM) was added to RPT to activate membrane purinoceptors and cause the release of ER Ca2+ and a transient increase in Ca2+f levels. A 10 min pretreatment with 2 μM U73122 inhibited the ATP-induced ER Ca2+ release by 76% but had no effect on thapsigargin-induced ER Ca2+ release (data not shown). These results suggest that antimycin A-induced ER Ca2+ release is through a PLC-independent mechanism.
Examination of store-operated Ca2+ entry in rabbit RPT suspensions
To determine whether the increase in Ca2+f following antimycin A addition was partly due to extracellular Ca2+ influx, 900 μM EGTA was added immediately prior to antimycin A and Ca2+f levels determined between 60 and 90 s later. Chelation of extracellular Ca2+ significantly reduced the antimycin A-induced Ca2+f increase from 182±5 to 109±10 nM. Extracellular Ca2+ influx was verified using a Mn2+ quench experiment, in which extracellular Mn2+ enters a cell similarly to Ca2+ but quenches the fluorescence of Fura.29 After exposure to 10 μM antimycin A in the presence of 50 μM Mn2+, Fura fluorescence was partially quenched, verifying that some extracellular Ca2+ influx occurred immediately after exposure to antimycin A (data not shown).
These experiments suggested that store operated Ca2+ entry may occur during RPT injury. To explore this possibility, control experiments were performed in which RPT in 1 mM extracellular Ca2+ were exposed to diluent (dimethylsulfoxide), thapsigargin, or antimycin A for 2 min before the addition of 10 mM Ca2+. Addition of 10 mM Ca2+ after DMSO, thapsigargin and antimycin A resulted in Ca2+f increases of 11, 56 and 10 nM, respectively. Although not typical of the large increases observed in other models,11,30 a reproducible small increase in Ca2+f levels was observed following addition of 10 mM Ca2+ in thapsigargin-treated versus diluent-treated RPT. However, the addition of 10 mM Ca2+ following 10 μM antimycin A exposure did not result in significant Ca2+f increases, suggesting that store operated Ca2+ entry does not occur following mitochondrial inhibition and ATP depletion.
Effect of thapsigargin and antimycin A on calpain activity
To determine whether ER Ca2+ release can increase calpain activity, thapsigargin was added to RPT and calpain substrate hydrolysis measured. Thapsigargin 5 μM induced a significant increase in substrate hydrolysis within 2 min of addition, indicating that ER Ca2+ release increases calpain activity (Figure 6A). Antimycin A increased calpain activity numerically in a concentration-dependent manner with 10 μM antimycin A increasing calpain activity 25% in the first 2 min after its addition, in the same time frame as the ER Ca2+ release. (Figure 6B). Inhibition of calpain activity with calpain inhibitor 1 (300 μM) prevented the antimycin A-induced increase in calpain activity (data not shown).

The effect of thapsigargin (Thaps) and antimycin A (AA) on calpain activity. After an initial baseline reading, Thaps (5 μM) or AA was added and readings were continued an additional 2 min. Bars represent the percentage of control calpain activity during the first 2 min after addition of Thaps or AA. Bars represent means±s.e. (n=3–4). Bars with different letters are significantly different from one another (P<0.05)
Effect of ER Ca2+ depletion on antimycin A- and TFEC-induced cell death
A preliminary study demonstrated that thapsigargin-induced ER Ca2+ release prior to antimycin A exposure blocked RPTC cell death.12 Additional experiments were conducted to further characterize the ER Ca2+ depletion and cell death induced by antimycin A and TFEC. Treatment of RPT with 10 μM antimycin A for 30 min resulted in an increase in LDH release (Figure 7A). A 5-min pretreatment with 5 μM thapsigargin or 100 μM cyclopiazonic acid prevented antimycin A-induced LDH release. Thapsigargin and cyclopiazonic acid pretreatment also decreased LDH release induced by a 3 h exposure to TFEC (Figure 7B). To document that the cytoprotective effect required ER Ca2+ depletion prior to toxicant exposure, thapsigargin was added after antimycin A or TFEC and LDH release measured. The addition of thapsigargin 15 min after antimycin A or 1 h after TFEC had no effect on antimycin A- or TFEC-induced LDH release (Figure 7).

(A) The effects of thapsigargin (Pre-thaps or Post-thaps) and cyclopiazonic acid (CPA) on antimycin A (AA)-induced LDH release. Thapsigargin (5 μM) was added 5 min prior to AA (Pre-thaps) or 15 min after AA (Post-thaps). CPA (100 μM) was added 5 min prior to AA. LDH release was determined 30 min after AA addition. (B) The effect of thapsigargin (Pre-thaps or Post-thaps) and CPA on tetrafluoroethyl-L-cysteine (TFEC)-induced LDH release. Thapsigargin (5 μM) was added 5 min prior to TFEC (Pre-thaps) or 60 min after TFEC (Post-thaps). CPA (100 μM) was added 5 min prior to TFEC. LDH release was determined 180 min after TFEC addition. Bars represent means±s.e. (n=3–4). Bars with different letters are significantly different from one another (P<0.05)
Discussion
Increases in Ca2+f have been demonstrated to play a critical role during oncosis in numerous models. For example, a study using concurrent measurement of Ca2+f and cell viability with propidium iodide revealed that increases in Ca2+f occur prior to plasma membrane damage in hypoxia-treated RPT.3 In addition, in RPT exposed to antimycin A for 30 min, chelation of intracellular and extracellular Ca2+, as well as the Ca2+ channel blocker nifedipine, inhibited Ca2+ uptake and were cytoprotective.7 Depletion of ER Ca2+ stores prior to antimycin A exposure or hypoxia prevented RPT cell death, indicating the important role of ER Ca2+.12 This study suggested that ER Ca2+ release, increased Ca2+f levels and extracellular Ca2+ influx are important mediators of oncosis. Further, the observation that inhibitors of the Ca2+-activated protease calpain are cytoprotective and block extracellular Ca2+ influx in RPT exposed to diverse toxicants suggest that calpains are a downstream target of elevated Ca2+f.7,20,21 These studies resulted in the model illustrated in Figure 1.
In the first series of experiments, antimycin A produced a rapid and sustained increase in Ca2+f. The relative increase in Ca2+f was similar to that produced by known inhibitors of the ER Ca2+-ATPase, thapsigargin and cyclopiazonic acid. Further, neither the addition of thapsigargin prior to antimycin A nor the addition of antimycin A prior to thapsigargin resulted in an increase in Ca2+f when the second agent was added. These results show that the increase in Ca2+f observed following antimycin A exposure is due to ER Ca2+ release. Thapsigargin and cyclopiazonic acid produced a transient increase in Ca2+f since ATP is present and the plasma membrane Ca2+-ATPase decreases Ca2+f. In contrast, the sustained increase in Ca2+f observed following antimycin A exposure is likely due to the absence of ATP and the inability of the plasma membrane Ca2+-ATPase to pump Ca2+ out of the cell. In addition, antimycin A-induced Ca2+ influx is likely to contribute to the sustained Ca2+ rise.
Emptying of intracellular Ca2+ stores has been linked to store operated Ca2+ entry across the plasma membrane in numerous cell types.31 For example, in a study by Demaurex et al.,32 cyclopiazonic acid treatment of HL-60 cells in the presence of extracellular Ca2+ resulted in a sustained increase in Ca2+f, indicating a Ca2+ influx pathway secondary to ER Ca2+ release. A Ca2+ influx pathway also was observed in MDCK cells, accounting for half of the thapsigargin-evoked Ca2+ signal.30 The data in the present manuscript suggest that store operated Ca2+ entry may occur to a limited degree in freshly-isolated rabbit RPT upon ER Ca2+ store depletion with thapsigargin, but does not occur during antimycin A-induced cell injury and death. These findings indicate the absence of store operated Ca2+ entry during injury and support prior evidence demonstrating an ATP requirement for store operated Ca2+ entry.33
In the second series of experiments we determined whether the antimycin A-induced ER Ca2+ release was the result of ATP depletion or PLC/IP3 pathway. Pretreatment with the phospholipase C inhibitor U73122 had no effect on the antimycin A-induced Ca2+f increase, suggesting that the PLC/IP3 pathway is not responsible for the observed ER Ca2+ release. However, antimycin A produced a rapid fall in ATP levels in the time frame of ER Ca2+ release. Within 1 min ATP depletion was 96% in RPT treated with 10 μM antimycin A. This degree of ATP depletion corresponds to an intracellular ATP concentration of approximately 150 μM (using the data in Figure 4 and an intracellular water volume of 2.4 μl/mg protein). Since the reported Km for ATP on the ER Ca2+-ATPase is 50–300 μM,34 the ATP depletion produced by antimycin A would result in inhibition of ER Ca2+-ATPase activity within the time frame of ER Ca2+ release. Further, this level of ATP depletion also would inhibit the plasma membrane Ca2+-ATPase activity, resulting in the sustained Ca2+f increase.
ATP depletion and Ca2+f increases both occurred within 1 min following antimycin A exposure. The trend of ATP depletion suggests that Ca2+f would begin to increase when ATP depletion exceeds 80 to 85%. A number of studies over the past 15–20 years have noted that extensive ATP depletion is required for the initiation of oncosis. For example, Lieberthal et al.1 found that oncosis occurred in cultured mouse RPT if ATP was depleted greater than 85%. Our results are consistent with their observations and provide an explanation relating ATP depletion to ER Ca2+-ATPase inhibition, ER Ca2+ release and the onset of oncosis.
We have shown that diverse calpain inhibitors are cytoprotective and that calpain inhibition blocks antimycin A-induced extracellular Ca2+ influx.7,21 These studies suggested that for calpains to be activated, Ca2+f must increase from an intracellular source. ER Ca2+ release produced by thapsigargin or antimycin A resulted in a rapid increase in calpain activity. Thus, antimycin A-induced ATP depletion inhibits the ER Ca2+-ATPase, releasing the ER Ca2+ store and resulting in increases in Ca2+f and calpain activity. The increase in calpain activity following thapsigargin exposure does not cause oncosis because ATP levels are maintained. Calpains have a number of substrates, including cytoskeletal proteins, which may mediate the cell injury/death process. The identification of these substrates is the next step in understanding the mechanism of oncosis.
We extended our previous findings12 and determined whether ER Ca2+ depletion protects RPT from TFEC toxicity. TFEC is a nephrotoxicant that is metabolized in RPT to a reactive electrophile that is capable of binding to nucleophiles and producing oncosis.35 Pretreatments with thapsigargin or CPA were protective against antimycin A- and TFEC-induced cell death. However, thapsigargin did not alter LDH release if given after the initial exposure to the toxicants. Since oncosis produced by diverse mechanisms (reactive electrophile, mitochondrial inhibition, hypoxia) is blocked by ER Ca2+ depletion, ER Ca2+ signaling appears to be critical for oncosis.
From these data, we suggest that during ischemic/toxicant induced injury, rapid ATP depletion results in ER Ca2+ release, extracellular Ca2+ influx and a sustained increase in Ca2+f. The sustained increase in Ca2+f activates calpains to mediate the late stage cell death events, including a second phase of extracellular Ca2+ influx, cell swelling and the loss of plasma membrane integrity (Figure 1).
Materials and Methods
Reagents
Antimycin A, dimethylsulfoxide, thapsigargin and cyclopiazonic acid were purchased from Sigma Chemical Co. (St. Louis, MO, USA). U73122 and Fura-PE3-2AM were obtained from Biomol (Plymouth Meeting, PA, USA) and TEFLABS (Austin, TX, USA), respectively. Tetrafluoroethyl-L-cysteine (TFEC) was synthesized according to the method of Moore and Green36 and was a gift from Dr. Edward A Lock (Zeneca, Cheshire, UK). The sources of the remaining chemicals have been reported previously37,38 or were obtained from Sigma Chemical Co. All glassware was silanized and autoclaved prior to use. All media were sterilized by filtering prior to use.
Preparation and incubation of RPT
Rabbit RPT were isolated and purified by the method of Rodeheaver et al.37 and suspended in an incubation buffer containing 1 mM alanine, 4 mM dextrose, 2 mM heptanoate, 4 mM lactate, 5 mM malate, 115 mM NaCl, 15 mM NaHCO3, 5 mM KCl, 2 mM NaH2PO4, 1 mM MgSO4, 1 mM CaCl2 and 10 mM HEPES (pH 7.4, 295 mOsm/kg). RPT suspensions (2 mg cellular protein/ml) were incubated at 37°C in an orbital shaking water bath (180 r.p.m.) under 95% air/5% CO2 (40 ml/min flow rate). All experiments contained a 15-min preincubation period prior to any experimental manipulations.
Measurement of Ca2+f
RPT suspensions were incubated with 2 μM Fura-PE3-2AM and 0.1% pluronic acid for 60 min at 25°C. Fura-PE3-2AM resists the rapid leakage and compartmentalization seen with Fura-2AM, yet has similar spectral properties.39,40 RPT were washed and resuspended in 37°C incubation buffer. Ca2+f was measured with a Hitachi F2000 spectrofluorometer (Hitachi Instruments, Danbury, CT, USA) equipped with a magnetic stirrer and a thermostatic cell holder (37°C). Following a 30 s equilibration period, baseline readings were obtained for 30 s. Antimycin A (0.3–10 μM), thapsigargin (5 μM), cyclopiazonic acid (100 μM), or the diluent (dimethylsulfoxide, <0.5% of total volume) was added and Ca2+f was monitored for 2 min. For calibration, EGTA (1 mM) was added to correct for any extracellular Fura-PE3-2AM. After adding back CaCl2 (2 mM), maximum and minimum fluorescence were determined using Triton X-100 (0.2%) followed by the Ca2+ chelator EGTA (20 mM). Readings were taken every 0.5 s and fluorescence measurements were made by alternating excitation wavelengths between 340 and 380 nm with continuous monitoring of emission at 510 nm. Ca2+f levels were determined using the following formula: [Ca2+]f=Kd(R-Rmin)/(Rmax-R)×(Fmin(380)/Fmax(380))41, where Kd=290 nM, R=the fluorescence emission ratio at 340 nm: 380 nm excitation and (Fmin(380)/Fmax(380)) is the ratio of minimum to maximum fluorescent intensity measured at 380 nm. For Mn2+ quench experiments, Mn2+ (50 μM) was added prior to antimycin A. Fluorescence was measured at the excitation wavelength of 360 nm and emission at 510 nm to achieve Ca2+-independent fluorescence.29
Biochemical assays
Release of LDH into the medium was used as a marker of cell death as described previously.42 ATP levels were analyzed by reverse-phase high performance liquid chromatography as described previously.37 Cellular calpain activity was measured as described previously21 with the following modifications. Baseline calpain activity was determined for 15 min after substrate (SLLVY-AMC) addition. Antimycin A or thapsigargin was then added and additional readings taken for the next two min. Calpain activity is reported as a percentage of the control linear change in fluorescence.
Statistics
Data are presented as means±s.e.m. of at least three experiments. RPT suspensions isolated from one rabbit represent a separate experiment (N of 1). Data were analyzed by ANOVA and multiple means compared using Student-Newman-Keuls test with P<0.05 indicating significance.
Abbreviations
- ER:
-
endoplasmic reticulum
- Ca2+f:
-
cytosolic free Ca2+; RPT, renal proximal tubule
- SERCA:
-
smooth endoplasmic reticulum Ca2+-ATPase; LDH, lactate dehydrogenase
- IP3:
-
inositiol trisphosphate
- PLC:
-
phospholipase C
References
Lieberthal W, Menza SA, Levine JS . 1998 Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells Am. J. Physiol. 274: F315–F327
Jiang T, Grant RL, Acosta D . 1993 A digitized fluorescence imaging study of intracellular free calcium, mitochondrial integrity and cytotoxicity in rat renal cells exposed to ionomycin, a calcium ionophore Toxicol. 85: 41–65
Kribben A, Wieder ED, Wetzels JF, Yu L, Gengara PE, Burke TJ, Schrier RW . 1994 Evidence for role of cytosolic free calcium in hypoxia-induced proximal tubule injury J. Clin. Invest. 93: 1922–1929
Takano T, Soltoff SP, Murdaugh S, Mandel LJ . 1985 Intracellular respiratory dysfunction and cell injury in short-term anoxia of rabbit renal proximal tubules J. Clin. Invest. 76: 2377–2384
Wetzels JF, Yu L, Wang X, Kribben A, Burke TJ, Schrier RW . 1993 Calcium modulation and cell injury in isolated rat proximal tubules J. Pharmacol. Exp. Ther. 267: 176–180
Rose UM, Bindels RJ, Jansen JW, Van Os CH . 1994 Effects of Ca2+ channel blockers, low Ca2+ medium and glycine on cell Ca2+ and injury in anoxic rabbit proximal tubules Kidney Int. 46: 223–229
Waters SL, Sarang SS, Wang KK, Schnellmann RG . 1997 Calpains mediate calcium and chloride influx during the late phase of cell injury J. Pharmacol. Exper. Ther. 283: 1177–1184
Almeida AR, Bunachak D, Burnier M, Wetzels JF, Burke TJ, Schrier RW . 1992 Time-dependent protective effects of calcium channel blockers on anoxia- and hypoxia-induced proximal tubule injury J. Pharmacol. Exper. Ther. 260: 526–532
Berridge MJ . 1993 Inositol trisphosphate and calcium signaling Nature 361: 315–325
Pozzan T, Rizzuto R, Volpe P, Meldolesi J . 1994 Molecular and cellular physiology of intracellular calcium stores Physiol. Rev. 74: 595–636
Bird GSJ, Putney JW . 1993 Inhibition of thapsigargin-induced calcium entry by microinjected guanine nucleotide analogues. Evidence for the involvement of a small G-protein in capacitative calcium entry J. Biol. Chem. 268: 21486–21488
Waters SL, Wong JK, Schnellmann RG . 1997 Depletion of endoplasmic reticulum calcium stores protects against hypoxia- and mitochondrial inhibitor-induced cellular injury and death Biochem. Biophys. Res. Comm. 240: 57–60
Bronk SF, Gores GJ . 1993 pH-dependent nonlysosomal proteolysis contributes to lethal anoxic injury of rat hepatocytes Am. J. Physiol. 264: G744–G751
Croall DE, Demartino GN . 1991 Calcium-activated neutral protease (calpain) system: structure, function and regulation Physiol. Reviews 71: 813–847
Edelstein CL, Wieder ED, Yaqoob MM, Gengaro PE, Burke TJ, Nemenoff RA, Schrier RW . 1995 The role of cysteine proteases in hypoxia-induced rat renal proximal tubular injury Proc. Natl. Acad. Sci. USA. 92: 7662–7666
Edelstein CL, Yaqoob MM, Schrier RW . 1996 The role of the calcium-dependent enzymes nitric oxide synthase and calpain in hypoxia-induced proximal tubule injury Renal Failure 18: 501–511
Nicotera P, Hartzell P, Baldi C, Svensson SA, Bellomo G, Orrenius S . 1986 Cystamine induces toxicity in hepatocytes through the elevation of cytosolic Ca2+ and the stimulation of a nonlysosomal proteolytic system J. Biol. Chem. 261: 14628–14635
McDonald MC, Mota-Filipe H, Paul A, Cuzzocrea S, Abdelrahman M, Harwood S, Plevin R, Chatterjee PK, Yaqoob MM, Thiemermann C . 2001 Calpain inhibitor I reduces the activation of nuclear factor-kappaB and organ injury/dysfunction in hemorrhagic shock FASEB J. 15: 171–186
Wang KK, Yuen PW . 1994 Calpain inhibition: an overview of its therapeutic potential Trends Pharmacol. Sci. 15: 412–419
Schnellmann RG, Yang X, Cross TJ . 1994 Calpains play a critical role in renal proximal tubule (RPT) cell death. Canad J. Physiol. Pharmacol. 72: 602
Harriman JF, Waters-Williams S, Chu D-L, Powers JC, Schnellmann RG . 2000 Efficacy of novel calpain inhibitors in preventing renal cell death J. Pharmacol. Exp. Ther. 294: 1083–1087
Wictome M, Michelangeli F, Lee AG, East M . 1991 The inhibitors thapsigargin and 2,5-di(tert-butyl)-1,4-benzohydroquinone favour the E2 form of the Ca2+,Mg(2+)-ATPase FEBS Lett. 304: 109–113
Zachetti D, Clementi E, Fasolato C, Lorenzon P, Zottini M, Grohovaz F, Fumagalli G, Pozzan T, Meldolesi J . 1991 Intracellular Ca2+ pools in PC12 cells. A unique, rapidly exchanging pool is sensitive to both inositol 1,4,5-trisphosphate and caffeine-ryanodine J. Biol. Chem. 266: 20152–20158
Gullans SR, Brazy PC, Soltoff SP, Dennis VW, Mandel LJ . 1982 Metabolic inhibitors: effects on metabolism and transport in the proximal tubule Am. J. Physiol. 243: F133–F140
Berridge MJ . 1997 Elementary and global aspects of calcium signaling J. Physiol. 499: 291–306
Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, Bunting S . 1990 Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils J. Pharmacol. Exp. Ther. 255: 756–768
Thompson AK, Mostafapour SP, Denlinger LC, Bleasdale JE, Fisher SK . 1991 The aminosteroid U-73122 inhibits muscarinic receptor sequestration and phosphoinositide hydrolysis in SK-N-SH neuroblastoma cells. A role for Gp in receptor compartmentation J. Biol. Chem. 266: 23856–23862
Jan CR, Ho CM, Wu SN, Tseng CJ . 1998 The phospholipase C inhibitor U73122 increases cytosolic calcium in MDCK cells by activating calcium influx and releasing stored calcium Life Sci. 63: 895–908
Merrit JE, Jacob R, Hallam TJ . 1989 Use of manganese to discriminate between calcium influx and mobilization from internal stores in stimulated human neutrophils J. Biol. Chem. 264: 1522–1527
Jan CR, Ho CM, Wu SN, Tseng CJ . 1999 Mechanism of rise and decay of thapsigargin-evoked calcium signals in MDCK cells Life Sci. 64: 259–267
Putney JW Jr . 1997 Type 3 inositol 1,4,5-trisphosphate receptor and capacitative calcium entry Cell Calcium 21: 257–261
Demaurex N, Lew DP, Krause KH . 1992 Cyclopiazonic acid depletes intracellular Ca2+ stores and activates an influx pathway for divalent cations in HL-60 cells J. Biol. Chem. 267: 2318–2324
Marriott I, Mason MJ . 1995 ATP depletion inhibits capacitative Ca2+ entry in rat thymic lymphocytes Am. J. Physiol. 269: 766–774
Engelender S, DeMeis L . 1996 Pharmacological differentiation between intracellular calcium pump isoforms Mol. Pharmacol. 50: 1243–1252
Groves CE, Hayden PJ, Lock EA, Schnellmann RG . 1993 Differential cellular effects in the toxicity of haloalkene and haloalkane cysteine conjugates to rabbit renal proximal tubules J. Biochem. Toxicol. 8: 49–56
Moore RB, Green T . 1988 The synthesis of nephrotoxin conjugates of glutathione and cysteine Toxicol. Environ. Chem. 17: 153–162
Rodeheaver DP, Aleo MD, Schnellmann RG . 1990 Differences in enzymatic and mechanical isolated rabbit renal proximal tubules: comparison in long-term incubation In Vitro Cell Dev. Biol. 26: 898–904
Groves CE, Schnellman RG . 1996 Suspensions of rabbit renal proximal tubules In: Methods in Renal Toxicology Zalups RK and Lash LH, eds Boca Raton, FL: CRC Press pp. 147–162
Abe F, Mitsui M, Karaki H, Endoh M . 1995 Calcium compartments in vascular smooth muscle cells as detected by aequorin signal Br. J. Pharmacol. 116: 3000–3004
Vorndran C, Minta A, Poenie M . 1995 New fluorescent calcium indicators designed for cytosolic retention or measuring calcium near membranes Biophys. J. 69: 2112–2124
Grynkiewicz G, Poenie M, Tsien RY . 1985 A new generation of Ca2+ indicators with greatly improved fluorescence properties J. Biol. Chem. 260: 3440–3450
Moran JH, Schnellmann RG . 1996 A rapid beta-NADH-linked fluorescence assay for lactate dehydrogenase in cellular death J. Pharmacol. Toxicol. Meth. 36: 41–44
Acknowledgements
We would like to thank Dr. Phil Mayeux for the use of the spectrofluorophotometer during this study. We would also like to thank Dr. Brian Cummings for his review of the manuscript. This work was supported in part by NIH ES-09129 (RG Schnellmann) and American Heart Association/Heartland Affiliate predoctoral fellowships to JF Harriman and X Liu.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by T Ferguson
Rights and permissions
About this article
Cite this article
Harriman, J., Liu, X., Aleo, M. et al. Endoplasmic reticulum Ca2+ signaling and calpains mediate renal cell death. Cell Death Differ 9, 734–741 (2002). https://doi.org/10.1038/sj.cdd.4401029
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401029
Keywords
This article is cited by
-
Calcium receptor signaling and citrate transport
Urolithiasis (2018)
-
Heat stress induced apoptosis is triggered by transcription-independent p53, Ca2+ dyshomeostasis and the subsequent Bax mitochondrial translocation
Scientific Reports (2015)
-
Zerumbone, a ginger sesquiterpene, induces apoptosis and autophagy in human hormone-refractory prostate cancers through tubulin binding and crosstalk between endoplasmic reticulum stress and mitochondrial insult
Naunyn-Schmiedeberg's Archives of Pharmacology (2015)
-
Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells
Apoptosis (2010)
-
Calpain small-1 modulates Akt/FoxO3A signaling and apoptosis through PP2A
Oncogene (2009)
