
Abstract
Knowledge of the emerging pathways of cell death downstream of the p53 tumor suppressor and the TRAIL death-inducing ligand is suggesting ways to improve therapeutic design in cancer. In contrast to its unique G1 cell cycle arresting mechanism that is maintained by p21WAF1, there are signals transduced by p53 to multiple apoptotic effectors perhaps due to the importance of apoptosis in suppressing tumors. There is evidence for cytoplasmic as well as mitochondrial activation of caspases downstream of p53, although in some cell lineages the signal ultimately involves the mitochondria. The TRAIL signaling pathway appears promising for therapeutic development despite sharing some similarities with the toxic Fas and TNF pathways, in terms of effector molecules and downstream signals. One of the key findings is the tissue specificity of cell death responses, a feature that could be exploited in strategies to widen the therapeutic window of combination cancer therapies. Efforts continue to develop p53-targeted cancer therapy, and novel clues to enhance or block specific effectors may improve therapeutic design. Cell Death and Differentiation (2001) 8, 1066–1075
Similar content being viewed by others


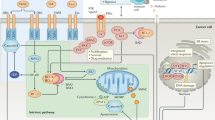
Introduction
One of the goals of the discipline of Molecular Oncology is to develop novel rational therapeutic strategies based on the specific molecular defects in human cancer. In the effort to develop novel therapies, one needs to identify targets that are different between normal cells and cancer cells. While microarrays provide a tool to discover specific differences between cancer and normal cells, as well as specific alterations in a given patient's tumor, p53 is a known target which is frequently overexpressed in mutant form in human cancer. Because p53 is also the most commonly mutated gene in human cancer and the p53 pathway is involved in the vast majority of tumors without mutations in p53, this p53 protein becomes an ideal target for therapeutic development in cancer. p53 abnormalities represent a fundamental difference between cancer cells and normal cells and as such they can be exploited in therapeutic design. Besides being an ideal target, p53 and its ability to induce tumor growth suppression through cell cycle arrest and the induction of apoptosis has been implicated in the cellular response to DNA damaging radiotherapy and chemotherapy.1,2 The emerging understanding of the pathways upstream and downstream of p53, and their signal- or tissue-specificity is providing a basis to develop more effective therapies. In addition to targeting the p53 pathway, there are emerging strategies that target the p53 molecule itself. These include small molecules that can inhibit wild-type p53 function or other agents that are capable of restoring wild-type p53 function in human cancer cells with mutant p53 protein.3,4 Yet other strategies are targeting key protein–protein interactions of p53 that disrupt its function in cancer.5,6,7
The TRAIL ligand and its signaling pathway is of interest for cancer therapy for a number of reasons. There are few agents that are truly cancer cell specific in terms of efficacy or cell death induction. TRAIL is an example of a molecule which specifically kills transformed and cancer cells but not most normal cells.8 This property has stimulated much work to understand the difference between the effect of TRAIL towards normal versus cancer cells, as well as efforts to understand defects in TRAIL signaling in cancers that fail to respond to its cytotoxic effects. One of the TRAIL receptors, KILLER/DR5, has been implicated in the cellular response to DNA damaging radiation or chemotherapy as a target of p53.9 Thus, although one of the attractive features of TRAIL is its ability to kill cancers with mutations in the p53 gene, the combination of TRAIL with chemotherapeutic agents has been found to be particularly effective in killing cancers with wild-type p53, presumably through induction of KILLER/DR5 expression.10,11
In developing a successful cancer therapeutic strategy, it becomes useful to incorporate ways to increase death of cancer cells while keeping normal cells alive (Figure 1). Cancer chemotherapy and radiotherapy, although often effective, is well known to have side-effects which can be severe and often limit therapy. Thus even established regimens can be improved and as a beginning, one needs to understand the basis of the toxicity of the therapeutic agents, in addition to appreciating their potential efficacy. Strategies that enhance death of cancer cells, while having low toxicity towards normal cells are needed in order to deliver successful therapy with an acceptable therapeutic window.

Desirable strategy to achieve therapeutic benefit in cancer therapy. Agents which effectively kill cancer cells are useful to the extent that toxicity to normal cells is tolerated. It is clear that therapeutic benefit can be achieved by increasing apoptosis of cancer cells as well as by lowering toxicity to normal cells upon exposure to anticancer therapy
p53, cancer and therapy
The p53 gene is the most commonly altered gene in human cancer, being involved through mutation in over 50% of all of human cancers world-wide. It is now believed that the vast majority of the cancers without mutational inactivation of p53 have alterations in the p53 pathway.12 This can occur through the targeted degradation of p53 induced by MDM2 amplification or overexpression, through human papillomavirus types 16 or 18 E6 protein which also targets p53 for degradation through the cellular E6-AP, and through deletion of the ARF gene which is upstream of p53. Because of the frequency of involvement of the p53 pathway in human cancer, it becomes an ideal target for therapeutic development as a basic difference between normal cells and cancer cells. An important ongoing drug development strategy is using ONYX-015, an E1B-deleted adenovirus with the ability to selectively replicate in p53-deficient or ARF-deficient cells, but not normal cells is currently in Phase II clinical trials.13 p53 mutations are also inherited in the germline of affected members of most Li-Fraumeni cancer prone families. These individuals are predisposed to breast cancer, sarcomas, brain tumors and leukemias, at a young age, in addition to other more common tumor types such as colon or lung cancer. Recent work has identified CHK2 mutations in some Li-Fraumeni kindreds without p53 mutation, providing more evidence for the involvement of other components of the p53 pathway.14 Another familial cancer predisposing syndrome that targets the p53 pathway is Ataxia Telangiectasia, where individuals inherit mutations in the ATM gene, another upstream kinase that regulates p53 in the cellular DNA damage response pathway.15
Mice deficient in p53 develop normally but are highly predisposed to spontaneous tumors.16 The majority of p53 homozygous-null mice develop lymphomas and sarcomas before 6 months of age and most are dead by 9 months. The heterozygous mice also develop tumors with longer latency and with more occurrences of adenocarcinomas, although lymphomas and sarcomas are still the most common tumors. In an Rb-deficient background p53-null mice develop endocrine tumors and in a telomerase-null background they are predisposed to epithelial tumors with a variety of non-reciprocal translocations.17,18 p53 deficiency prolongs survival of MDM2-null mice to term and partially rescues BRCA1-null embryos.19,20,21
p53-deficient cells have been known for about a decade to harbor genomic instability, a feature also observed in older fibroblasts derived from Li-Fraumeni patients.22,23 A recent experiment has shed some light on the relationship of genomic instability and the tumor prone phenotype due to dysfunctional p53.24 The growth arrest and DNA damage-inducible gene GADD45, originally discovered as an upregulated transcript in UV-exposed cells is a downstream transcriptional target of p53.25 The generation of a GADD45 knockout mouse has allowed the examination of GADD45-null cells which, like p53-null cells, have genomic instability. However, the GADD45-null mice do not develop spontaneous tumors, unless they are also exposed to ionizing radiation. These experiments appear to have uncoupled genomic instability from tumor susceptibility, i.e. genomic instability is a contributing factor but is not sufficient for tumor development.
p53 is a potent tumor suppressor gene such that if added back into cancer cells it suppresses growth despite other molecular genetic changes in the cells.26,27 This is particularly relevant and important as a therapeutic strategy, for example in the gene therapy of cancer. p53 induces growth arrest and/or apoptosis either when exogenously administered or through its involvement in the cellular response to DNA damage.
In addition to clearly playing a role in cancer susceptibility, p53 appears to be a major determinant of sensitivity of cells to chemo- and radiotherapy.28 Some of the evidence has come from studies of p53-null mouse thymocytes which are radio-resistant unlike thymocytes expressing wild-type p53.29 p53-null fibroblasts that also express E1A and ras oncogenes are also resistant not only to ionizing radiation, but also to a number of DNA damaging chemotherapeutic agents.30 In the mouse cells the presence of E1A and ras confers to the wild-type fibroblasts sensitivity to DNA damaging agents. Additional evidence for the role of p53 in chemo- and radio-sensitivity has come from the NCI cell screen.31 While we are still learning about the complex gene expression profiles, one of the early results was that the vast majority of clinically useful agents are most effective in killing wild-type p53-expressing cells. It is important to point out that some data suggests that p53 may be important as an early event in cell fate, but that eventually especially at higher doses both p53 wild-type and p53-null cells are killed by radiation through ‘reproductive death’. Such criticisms are important to note in deciding which assay one uses to determine the effect of a particular therapy.32,33,34 The early difference in sensitivity between wild-type and mutant p53-expressing cells offers an opportunity to develop more effective therapies. Based on the results from the NCI cell screen, there are efforts to screen for agents which can selectively kill p53-deficient cells. Taxol is an example of one such agent which has been in clinical use for over a decade, and was recognized later in the cell screen as more likely to kill p53-deficient cells.31 There are other compounds that are known to more efficiently or to preferentially kill p53-deficient cells and some of them have even gone to clinical trials but failed because of toxicity.31 The hope is that some of these agents could be revisited and modified in ways to reduce toxicity while maintaining specificity for p53-mutant or p53-null cells.31
Regulation, structure, and signals downstream of p53
Clear and distinct signaling pathways are emerging upstream of p53 stabilization in response to a number of stresses.35,36 There appear to be at least three or more main pathways upstream of p53.1,37 The now classical DNA damage response pathway involves kinases that become ‘activated’ through unclear mechanisms upon cellular exposure to DNA damaging agents such as ionizing radiation or topoisomerase II inhibitors (Figure 2). These DNA damaging agents cause breaks in the cellular DNA which may be an initiating signal. Once ‘activated’ the kinases ATM or CHK2 can each phosphorylate p53 on N-terminal residues within its transactivation domain thereby leading to increased p53 activity. Specifically, physical association between p53 and ATM leads to phosphorylation on serine 15 of p53 which is now believed to enhance its transcriptional activity rather than leading to an increase in its half-life per se.38 ATM also acts on CHK2 to phosphorylate it.15 CHK2 in turn phosphorylates p53 on serine 15 which disrupts its binding to its negative regulator MDM2 which is an E3 ubiquitin ligase. Release of MDM2 from p53 leads to p53 protein stabilization which in turn allows p53 to mediate its potent downstream effect on cell growth suppression.37 DNA damage initiated signals leading to activation of ATM and CHK2 have recently been recognized to also target the phosphorylation of MDM2 further inhibiting MDM2:p53 interaction.39 In addition to targeting p53 for ubiquitin-mediated proteolysis, and directly interacting with and physically blocking the transactivation domain of p53, MDM2 interaction also targets p53 for nuclear export thereby also facilitating its degradation and inhibiting its effects on gene expression in the nucleus.40 It is of interest that the degradation of p53 by MDM2 utilizes different structural requirements within p53 as compared to the human papillomavirus E6-targeted degradation.41

Cell cycle arrest mediated by p53 targets and other DNA damage checkpoint proteins. In G1 phase, exposure of cells to DNA damaging ionizing radiation leads to rapid degradation of cyclin D1 and cdc25a. A slower transcriptional activation of p21WAF1 leads to inhibition of cyclin-dependent kinase activity, Rb hypophosphorylation and growth arrest of cells in G1. In G2, DNA damage leads to inactivation of cdc25 proteins through several mechanisms that ultimately arrest cell growth. In addition, transcriptional control of p53 target genes such as GADD45, 14-3-3σ, and p21WAF1 serves to maintain a G2 arrest. ATM and ATR also regulate S-phase checkpoints in response to DNA damage. p53 is regulated by other proteins such as MDM2 and p14ARF which ultimately control its stability and nuclear localization. p53 controls other transcriptional targets that lead to apoptosis or growth inhibition
A second pathway involving the ARF protein appears to constitute the cellular response to inappropriate growth signals, the so-called ‘oncogene checkpoint’. For over two decades it has been observed that p53 protein levels are high in transformed cells. For example, p53 was originally isolated as a highly expressed protein that bound to SV40 large T-antigen in transformed cells or as an aberrantly expressed ‘tumor antigen’. It was also known that a number of cellular and other viral oncogenes can increase the expression level of cellular p53. These include c-Myc, ras, and adenovirus E1A proteins. It has recently become clear that ARF, the alternative-reading frame at the INK4A locus, is a protein that interacts with MDM2 thereby sequestering it in nucleolar structures away from p53 which can then become more stable.36 There is recent evidence that ARF has p53-independent, MDM2-dependent effects on growth suppression through an as yet unclear mechanism.42 There is also evidence for MDM2-independent nuclear export of p53 following DNA damage through a newly discovered nuclear export signal in the amino-terminal region of p53.43
A third pathway is triggered by exposure to UV radiation. This UV-initiated p53 stabilization does not require ATM, but may utilize a related kinase called ATR.44,45 DNA replication blockade also triggers p53 stabilization in an ATM-independent manner.46,47 The ultimate effect of the signals upstream of p53 appear to lead to release of MDM2 from p53. This is the basis of a drug development strategy whose aim is to deliver peptides or small molecules into cells that still have wild-type p53 but which may overexpress MDM2. Such peptides which may fit into the p53:MDM2 interacting groove may and displace MDM2 thereby stabilizing the tumor suppressor p53.48
Not all upstream signals target the N-terminus and MDM2 binding. It is known that ATM can also activate an unknown phosphatase that dephosphorylates p53 near the C-terminus, thereby creating a 14-3-3 binding site that appears to enhance DNA binding activity.49 It is also known that C-terminal acetylation events can activate DNA binding by p53.50 Yet another pathway leading to p53 activation may involve phosphorylation at serine 46 which may result in a p53 protein more likely to induce cell death through target gene selectivity, i.e. p53AIP1.51 Acetylation and apoptosis-specific phosphorylation events may provide other targets for drug development.
The open reading frame of wild-type p53 encodes a 393 amino acid polypeptide with a central DNA binding domain between aa 100–300.37 The N-terminal region (aa 1–42) encodes a potent transactivation domain, and the C-terminal region encodes an oligomerization domain, a nuclear localization signal, acetylation, phosphorylation and dephosphorylation sites and a domain that can regulate DNA binding. Most tumor-derived hotspots in p53 are within its DNA binding domain. In fact one of the most elegant results of the past decade was the demonstration with the crystal structure of the DNA binding domain that several of the hotspots in human cancer are residues that normally directly contact the p53 DNA-binding consensus response element.52 Such evidence has supported the thesis that the sequence-specific DNA-binding property of p53 protein is critical for its function as a tumor suppressor. There is an alternatively spliced form of p53 and it is now known that there are two other mammalian homologues of p53.53 These homologues are p63 and p73. Alternatively spliced forms of p63 may be aberrantly overexpressed in some tumors and p73 may be silenced in other tumors.54,55 The evidence thus far is weak, indicating that either p63 or p73 are tumor suppressor genes. However, there is a growing literature on interactions between p73 and tumor-derived mutant p53, as well as recent evidence that p73 may be regulated by E2F1, a protein that is involved in p53-independent DNA damage-induced cell death.56 As such, these observations provide clues for drug development strategies that may increase expression of p53 family members or enhance sequestration of mutated p53 by p53 family members. There appears to be only one p53 family member in Drosophila, and this homologue is most similar to p53 versus p63 or p73.57,58 It has been observed that HPV 16 or 18 E6 protein, which targets p53 for degradation in cervical cancers exposed to these pathogenic strains, does not target the p53 family member p73 for degradation.59 Such observations form the basis for a gene replacement therapeutic strategy relying on p73 to induce apoptosis in HPV(+) cervical cancers. There are other strategies that are aimed at targeting the E6/E6-AP-dependent pathway of p53 degradation.59
Once activated through phosphorylation, dephosphorylation, acetylation or protein–protein interactions, p53 acts as a tumor suppressor through effects on gene expression.1,37 There is a growing list of target genes, which are repressed by p53. Although the mechanism of repression is not yet clear, and appears not to be mediated through sequence-specific DNA binding (at least of its known target binding DNA sequence), several of the targets may be necessary to suppress in order for p53 to suppress growth or induce cell death. The clearest mechanisms downstream of p53 involve the direct upregulation of expression of target genes containing p53 DNA-binding consensus sites in their regulatory regions.1,37 In the case of cell cycle arrest, p53 transcriptionally upregulates p21WAF1, the universal cell cycle inhibitory protein which arrests cells primarily in the G1-phase (Figure 2). Recent data has shown that rapid p53-independent events following DNA damage involving degradation of cdc25a and cyclin D1 initiate G1 cell cycle arrest, whereas the transcriptional response by p53 to upregulate p21 is required for maintaining the cell cycle arrest.60 Although there are other targets of p53 implicated in G2 arrest, namely 14-3-3σ and GADD45, G1 arrest appears thus far to be the sole function of p21 (Figure 2). It is a unique feature of the cell cycle arrest response that one gene, p21, is uniquely upregulated and seemingly sufficient to induce G1 arrest. Although p21 is also induced in cells undergoing cell death, it appears not to induce cell death and the general belief at present is that p21 can protect cells from cell death caused by a variety of insults. p53 can induce expression of several proteins which are secreted including IGF-BP3, TSP1, and BAI1.1,37 The latter two are inhibitors of angiogenesis. With regard to apoptosis, the picture that is emerging is complex and appears to involve the upregulation of a growing list of downstream target genes. The recent generation of a transactivation-deficient knock-in p53 mouse has provided some of the best evidence that the transcriptional activation property of p53 is essential for tumor suppression.61
p53 pathways of apoptosis and cancer therapy
The transcriptional targets upregulated by p53 which can in-turn induce apoptosis fall into at least three categories: death domain containing proteins including two proapoptotic death receptors, proteins that act at the levels of the mitochondria including two proapoptotic Bc12 family members and three inhibitors of antiapoptotic Bc12 family members, and a third group of proteins that lead to the generation of reactive oxygen species.1,37 One of the clear results thus far is that no one target of p53-dependent apoptosis can fully account for the loss of death phenotype observed in cells-deficient for p53 (Figure 3). Thus one of the important questions is why are there so many targets of p53 that can induce cell death? Are all these targets and pathways used each time p53 induces cell death? One scenario is there may be tissue specificity or signal specificity to the response. There may even be some built-in redundancy in the signaling pathway, because of its importance in suppressing cancer.

Pathways downstream of p53 that can cause cell death. In response to apoptotic signals, p53 protein is stabilized and activated resulting in transcriptional activation of multiple target genes that cause apoptosis of cells. These include death receptors such as Fas/APO1 or KILLER/DR5, or proteins that act more directly on caspase activation including Bax, Bak, Noxa, PUMA, AIP1, and APAF1. Death receptors contain extracellular cysteine-rich ligand-binding domains (CRD) and intracellular death domains (DD). Other targets including PIDD and PIG genes have been found to be activated by p53. PIG genes lead to the generation of reactive oxygen species (ROS) which are toxic to cells. The initiator caspases involved in p53 signaling of death include caspases 8 and 9, whereas the downstream executioner caspases include caspases 3, 6, and 7. Inhibitors of p53-dependent cell death include Bc1-XL and FLIP, which act on cytoplasmic or mitochondrial pathways of death activation
In considering the importance of targets of p53 in the cell death response, it has sometimes been argued that many targets may be ‘artifacts’, (of overexpression of p53). Such generalizations risk overlooking potentially very important targets, including ones that may ultimately prove to be key targets for drug development. It is clear that genes containing classical p53 DNA-binding consensus response elements are excellent candidates for regulation by p53. It also seems somewhat intuitively obvious that cells which expend energy and utilize building blocks to upregulate gene expression would not do so for no reason or in a wasteful manner. It is therefore not unreasonable to presume that if a cell upregulates a death-inducing gene in response to p53 stabilization or overexpression, such a gene may be involved in the death response. Such a gene may be necessary but not sufficient, or it may not even be required under certain conditions. p53 is a transcription factor that has had plenty of time to evolve from the Drosophila homologue to respond to a variety of signals and to potentially be subject to a number of modifications or cellular interactions that modulate its target gene selectivity. This ‘target gene selectivity’ hypothesis remains one of the best models to at least begin to explain the divergent phenotypes (e.g. arrest or apoptosis) observed in response to p53 activation. The property of p53 as a transcription factor, with the exception of the unique p53–p21 connection (which is as yet not defined in Drosophila), allows it to upregulate the necessary targets to achieve a desired response. Based on the degeneracy of the p53 response element there may be at a minimum several hundred bona fide p53 effector genes. Moreover, in considering targets of p53 that are directly upregulated, the most attractive effectors are ones with a plausible mechanism of action, i.e. targets which can be linked to the caspase machinery of cell death.
How does p53 cause cell death? There is very good evidence that caspase 9 and APAF1 are crucial late downstream effectors of the p53-regulated cell death response.62 Such studies suggest that at least in some cell types, e.g. embryonic fibroblasts that also express E1A and ras, the p53-dependent death signal ultimately travels through the mitochondria leading to formation of the apoptosome. Recent studies have identified certain melanoma tumor cell lines with reduced expression levels of APAF1, and showed that the re-expression of APAF1 by the use of 5′-azacytidine could resensitize cells to the apoptotic effects of DNA damaging chemotherapeutic agents.63 APAF1 has also been recently described as a p53 target gene. There is also good evidence that that p53 directly controls death-inducing genes that can directly promote cytochrome c release (proapoptotic Bcl2 family members Bax and Bak, and inhibitors of anti-apoptotic Bcl2 family members Noxa, p53AIP1 and PUMA), as well as death-inducing genes of the death receptor class (Fas and KILLER/DR5; PIDD is a death-domain containing protein but which does not appear to be a receptor).1,9,37,51,64,65,66,67,68,69
A p53-dependent increase in death receptor expression is potentially a mechanism by which cells may be more effectively killed by the immune system, and it appears to be a mechanism for enhanced killing of cancer cells exposed to death-inducing ligands plus cytotoxic agents or radiation.9,70,72,73,74,75 Activation of death receptors such as Fas or KILLER/DR5 leads to recruitment of the adaptor FADD and the initiator caspases 8 and 10 to the cell membrane.8,76 Through induced proximity, caspases 8 and 10 become activated and can then trigger the caspase cascade. Downstream of caspase 8 is Bid, which can be cleaved and translocate to the mitochondria to trigger cytochrome c release through interactions with Bak or Bax. There is evidence that both caspases 8 and 9 are cleaved in response to p53 and that p53-dependent apoptosis can be blocked by inhibitors of caspase 8 or caspase 9, i.e. cFLIP or C8I, and BclXL or C9I, respectively.77,78 The available evidence thus far suggests that neither Fas nor Bax is required for p53-dependent cell death.79,80 However, because they are induced they may contribute to cell death under such situations. Bax appears to be important for neuronal cell death and the deficiency in cell death is much more pronounced if Bak is also deleted.68,81,82 It is noteworthy that Bax/Bak double-null mice, although deficient in DNA damage-induced cell death, do not develop tumors like the p53-null mice. There is recent evidence that suggests a preferential tissue-specific upregulation of KILLER/DR5 in the spleen and gastrointestinal tract, as compared to Fas and Bax.78 Such tissue-specific induction may be a clue that for a novel drug development strategy. Specifically, it would be expected if a particular effector of p53-dependent apoptosis is favored in a given tissue that targeting its inhibition may reduce toxicity in response to chemo- or radiotherapy. Such strategies are underway as the determinants of KILLER/DR5-induced death signaling and their loss in tumors is unraveled.83
The idea that blocking the p53 response to reduce toxicity is not novel.84 The novelty of the tissue-specific p53 response is to block specific targets favored in a particular tissue that is highly susceptible to toxicity. The potential target described here is the p53-favored KILLER/DR5 upregulation in the gut78 which is highly sensitive to radiation. Blocking KILLER/DR5 may not inhibit radiation-induced killing in other tissues but may reduce toxicity. This in only a hypothesis at present, and it is possible there may be other targets of p53 in gut that mediate toxicity or which may substitute for KILLER/DR5. None-the-less, current efforts to generically block p53 function do look promising in terms of reducing the toxicity of radiotherapy or chemotherapy. Use of the compound Pifithrin has allowed mice to survive a lethal dose of ionizing radiation without developing tumors.3 Recently, efforts have begun to be directed at developing a Pifithrin-containing cream that may block the undesired side-effect of chemotherapy leading to hair loss in cancer patients.85
Utilizing the TRAIL pathway in p53-targeted and p53-independent cancer therapy
Interest in the TRAIL pathway intersected with the p53 pathway in 1997 when the proapoptotic TRAIL receptor KILLER/DR5 was cloned from a subtractive hybridization screen as a DNA damage-inducible p53-regulated gene.8,86,87 Recent work has identified p53 response elements in the human genomic KILLER/DR5 locus, and the p53-dependent regulation of KILLER/DR5 appears to be conserved in the mouse.88,89 TRAIL is also of great interest to cancer biologists because of its ability to cause cell death of transformed and cancer cells, but not most normal cells8 (Figure 4). The resistance of normal cells to the cytotoxic effects of TRAIL appear to be in part mediated by high surface expression of TRAIL decoy receptors that compete for binding to the ligand and reduce activation of the death signal through the proapoptotic TRAIL receptors.10 Some normal cells also appear to express high levels of cellular FLIP, the Flice-inhibitory protein that is also overexpressed in some cancers.10 Recent work on elucidating the signaling pathway downstream of TRAIL receptors has provided good evidence for the involvement of the FADD adaptor and caspase 8 as the initiator caspase.90

Sensitivity and resistance to TRAIL-induced cell death. The cytotoxic ligand TRAIL kills cancer and transformed cells but not most normal cells. Normal cells are believed to be protected from TRAIL because they tend to express higher levels of TRAIL decoy receptors (TRID or TRUNDD). Most cancer cells are sensitive to TRAIL, but there are multiple mechanisms of resistance that have been described. Cancer cells can become resistant to TRAIL if they lose expression of the proapoptotic TRAIL receptors DR4 or KILLER/DR5. This can occur through homozygous deletion of DR4 or mutations in DR5. Inactivating DR5 mutations have been described in head and neck, lung and breast cancers. TRAIL resistance may develop if cells overexpress either FLIP or Bc1-XL. Neuroblastomas can become resistant to TRAIL through hypermethylation of caspase 8 which may be reversed by exposure to 5-aza-Cytidine. Chemotherapy or radiation can be combined with TRAIL to achieve synergistic cell killing, in part through p53-dependent upregulation of KILLER/DR5 expression
A number of studies have evaluated the sensitivity of human tumor cells to the cytotoxic effects of TRAIL. It is clear that not all cancer cells are sensitive to the killing effects of TRAIL.10 Correlations between loss of DR4 expression and elevated cellular FLIP expression as independent predictors of TRAIL resistance have been made in some studies.10 A recent study has identified hypermethylation of caspase 8 as a mechanism of inactivation in neuroblastomas leading to TRAIL resistance which could be reversed by exposing cells to the demethylating agent 5-azacytidine.91 Loss of DR4 expression through homozygous deletion resulting from translocation events was observed in the aneuploid FaDu nasopharyngeal cancer cell line.92 FaDu cells which are resistant to TRAIL were partially sensitized through exogenous expression of wild-type DR4. DR5 mutations have been described in head and neck, lung and breast cancer.93,94,95 A truncating mutation in the death domain was found in head and neck cancer and a number of mutations in the death domain were observed in lung and breast cancer, leading to loss of function. These studies support the notion that the proapoptotic TRAIL receptors are candidate tumor suppressor genes. Recent work has provided evidence for TRAIL production by endogenous natural killer cells in a pathway stimulated by γ-interferon.96 Blockade of TRAIL through the use of a blocking antibody was shown to lead to substantial effects on increasing tumor xenograft growth consistent with the idea that TRAIL may be a tumor suppressor in vivo. Further support for this pathway is suggested by experiments showing that the TRAIL receptor KILLER/DR5 is also upregulated in γ-IFN-treated cells.97 Yet other support for the idea that TRAIL receptors may be involved in suppressing tumors is suggested by a recent study that has identified a higher frequency of certain polymorphisms in the DR4 gene in the germline of individuals who developed cancer as compared to individuals without cancer.98
Preclinical studies utilizing TRAIL have provided in vivo evidence for exogenous recombinant TRAIL efficacy in suppressing tumor growth.99 Moreover, it appears that TRAIL efficacy can be increased in wild-type p53 expressing cells through the combined use of DNA damaging chemotherapeutics or ionizing radiation.8,70,71,72,73,74,75 This is believed to be due to the p53-dependent upregulation of the TRAIL receptor KILLER/DR5 following DNA damage. A direct test of this hypothesis was recently performed by showing inhibition of cell death due to the combination of p53 overexpression (delivered by adenovirus) and TRAIL through the use of a soluble extracellular domain of the KILLER/DR5 receptor to compete with TRAIL binding to the upregulated DR5.11 While it is clear that TRAIL is effective in killing mutant p53-expressing cancer cells and as such is a promising agent, the increased efficacy realized by activation of the p53 pathway may have therapeutic utility versus certain tumors. Other strategies under development include the combination of TRAIL with γ-IFN,96 or enhancement of cell killing through cell cycle modulation.100
Some studies have observed that recombinant human TRAIL may be toxic to some normal human or mouse cells.101,102 However, recent studies have suggested that not all preparations of TRAIL are equally toxic to normal human cells.103 It was reported that untagged full-length human TRAIL binds reversibly to liver cells whereas tagged-TRAIL which is crosslinked using an antibody binds irreversibly. The kinetics and molecular basis for these differences are still under investigation, as well as the ultimate impact of reversible versus irreversible TRAIL binding on cancer cell death in terms of efficacy. One study has provided evidence for cell-type specific effects of TRAIL in killing normal or cancer cells.77 It was suggested that a potential approach to reduce the toxicity of TRAIL towards some normal cells may employ caspase 9 inhibitors which appear to protect Type II cells while permitting death of Type I cells.77 Such strategies may have a role in the design of trials for systemic use of TRAIL, as they allow the killing of Type I cancer cells while protecting normal cells that appear to die by a Type II mechanism. The prolongation of survival of Type II cells through the use of caspase 9 inhibitors was shown in long-term assays, suggesting that a brief exposure to TRAIL may not lead to irreversible events at the level of the mitochondria. The current evidence does support the idea that like Fas, TRAIL may kill cells by alternative pathways, i.e. Type I or Type II. Further studies on the molecular basis for these differences may provide novel targets for drug development.
Pharmacological restoration of p53 function in mutant p53-expressing cells
Screening for agents that could restore p53 function in cells with mutated p53 led to the identification of a class of bifunctional compounds that recover a wild-type epitope within the DNA binding domain of p53.4 The lead compound, CP-31398, was shown to restore transcriptional activity to mutant p53 transfected into p53-null cells.4 CP-31398 was shown to inhibit tumor xenograft growth in mice without appreciable toxicity.
Further studies of this compound are ongoing, but preliminary results indicate that most cancer cells exposed to CP-31398 undergo apoptosis.104 A minority of cell lines undergo cell cycle arrest prior to the apoptosis, or cell cycle arrest exclusively. Interestingly, CP-31398 appears to stabilize wild-type p53 protein, at higher doses.104 This drug appears to alter global gene expression patterns in a manner suggestive of effects on p53 targets as well as targets not known to be regulated by p53 or DNA damage. One proposed future direction might involve performing array screens with derivative compounds in an attempt to improve the specificity for mutant p53. This class of drugs is very interesting because of their unique mechanism of action and potential to treat cancers with mutant p53, and the potential exists for combining these agents with classical chemotherapies or TRAIL.
Conclusion and future prospects
Understanding the cell death pathways induced by p53 activation or engagement of death receptors by the TRAIL ligand is leading to novel strategies for therapeutic design in cancer. In particular, the emerging complex phenotypes involving cell- and tissue-specific signaling events is suggesting possible ways in which to shift the balance in favor of cancer cell death and against normal cell death to widen the therapeutic window. The p53 and TRAIL signaling pathways represent ideal targets for therapeutic development because of their relevance to cancer cell apoptosis and the numerous potential ways in which to intervene and modulate death responces and toxicity. Ultimately cancer therapy may become individualized at the molecular level, perhaps through the use of microarrays,105 and such approaches may identify specific targets in the death pathway whose induction or inhibition would improve therapeutic outcome.
Abbreviations
- TRAIL:
-
Tumor Necrosis Factor-related apoptosis-inducing ligand
- KILLER/DR5:
-
TRAIL Death receptor 5
References
Vogelstein B, Lane D, Levine AJ . 2000 Surfing the p53 network Nature 408: 307–310
Schmitt CA, Rosenthal CT, Lowe SW . 2000 Genetic analysis of chemoresistance in primary murine lymphomas Nature Med. 6: 1029–1035
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV . 1999 A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy Science 285: 1733–1737
Foster BA, Coffey HA, Morin MJ, Rastinejad F . 1999 Pharmacological rescue of mutant p53 conformation and function Science 286: 2507–2510
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP . 1996 Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain Science 274: 948–953
Stoll R, Renner C, Hansen S, Palme S, Klein C, Belling A, Zeslawski W, Kamionka M, Rehm T, Muhlhahn P, Schumacher R, Hesse F, Kaluza B, Voelter W, Engh RA, Holak TA . 2001 Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53 Biochemistry 40: 336–344
Beerheide W, Sim MM, Tan YJ, Bernard HU, Ting AE . 2000 Inactivation of the human papillomavirus-16 E6 oncoprotein by organic disulfides Bioorganic Medic. Chem. 8: 2549–2560
Ashkenazi A, Dixit VM . 1998 Death receptors: Signaling and modulation Science 281: 1305–1308
Wu GS, Kim K, El-Deiry WS . 2000 KILLER/DR5, a novel DNA-damage inducible death receptor gene, links the p53-tumor suppressor to caspase activation and apoptotic death Adv. Exp. Med. Biol. 465: 143–151
Kim KH, Fisher MJ, Xu SQ, El-Deiry WS . 2000 Molecular determinants of response to TRAIL in killing of normal and cancer cells Clin. Cancer Res. 6: 335–346
Kim K, Takimoto R, Dicker DT, Chen YH, Gazitt Y, El-Deiry WS . 2001 Enhanced TRAIL sensitivity by p53 overexpression in human cancer but not normal cell lines Int. J. Oncol. 18: 241–247
Vousden KH . 2000 p53: Death star Cell 103: 691–694
Nemunaitis J, Khuri F, Ganly I, Arseneau J, Posner M, Vokes E, Kuhn J, McCarty T, Landers S, Blackburn A, Romel L, Randlev B, Kaye S, Kirn D . 2001 Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer J. Clin. Oncol. 19: 289–298
Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DCR, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA . 1999 Science 286: 2528–2531
Kastan MB, Lim DS . 2000 The many substrates and functions of ATM Nature Rev. Mol. Cell. Biol. 1: 179–186
Ghebranious N, Donehower LA . 1998 Mouse models in tumor suppression Oncogene 17: 3385–3400
Harvey M, Vogel H, Lee EYHP, Bradley A, Donehower LA . 1995 Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin Cancer Res. 55: 1146–1151
Artandi SE, Change S, Lee SL, Alson S, Gottleib GJ, Chin L, DePinho RA . 2000 Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice Nature 406: 641–645
Jones SN, Roe AE, Donehower LA, Bradley A . 1995 Rescue of embryonic lethality in MDM2-deficient mice by absence of p53 Nature 378: 206–208
Hakem R, delaPompa JL, Eli A, Potter J, Mak TW . 1997 Partial rescue of Brca1(5-6) early embryonic lethality by p53 or p21 null mutation Nature Genet. 16: 298–302
Xu X, Qiao W, Linke SP, Cao L, Li W-M, Furth PA, Harris CC . 2001 Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis Nature Genet. 28: 266–271
Yin YX, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM . 1992 Wild-type p53 restores cell-cycle control and inhibits gene amplification in cells with mutant p53 alleles Cell 70: 937–948
Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD . 1992 Altered cell-cycle arrest and gene amplification potential accompanies loss of wild-type p53 Cell 70: 923–935
Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, Rosenberg MP, Zhan QM, Fernandez-Salguero PM, Morgan WF, Deng CX, Fornace AJ . 1999 Genomic instability in Gadd45a-deficient mice Nature Genet. 23: 176–184
Kastan MB, Zhan QM, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ . 1992 A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia telangiectasia Cell 71: 587–597
Baker SJ, Markowitz S, Fearon ER, Willson JKV, Vogelstein B . 1990 Suppression of human colorectal carcinoma cell growth by wild-type p53 Science 249: 912–915
Blagosklonny MV, El-Deiry WS . 1998 Acute overexpression of wt p53 facilitates anticancer drug-induced death of cancer and normal cells Int. J. Cancer 75: 933–940
Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T . 1994 p53 status and the efficacy of cancer therapy in-vivo Science 266: 807–810
Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T . 1993 p53 is required for radiation-induced apoptosis in mouse thymocytes Nature 362: 847–849
Lowe SW, Ruley HE, Jacks T, Housman DE . 1993 p53-dependent apoptosis modulates the cytotoxicity of anticancer agents Cell 74: 957–967
Weinstein JN, Myers TG, Oconnor PM, Friend SH, Fornace AJ, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, vanOsdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD . 1997 An information-intensive approach to the molecular pharmacology of cancer Science 275: 343–349
Brown JM, Wouters BG . 1999 Apoptosis, p53, and tumor cell sensitivity to anticancer agents Cancer Res. 59: 1391–1399
Waldman T, Zhang YG, Dillehay L, Yu J, Kinzler K, Vogelstein B, Williams J . 1997 Cell-cycle arrest versus cell death in cancer therapy Nature Med. 3: 1034–1036
Wu GS, El-Deiry WS . 1996 p53 and chemosensitivity Nature Med. 2: 255–256
Ryan KM, Phillips AC, Vousden KH . 2001 Regulation and function of the p53 tumor suppressor protein Curr. Opin. Cell Biol. 13: 332–337
Sherr CJ, Weber JD . 2000 The ARF/p53 pathway Curr. Opin. Genet. Dev. 10: 94–99
El-Deiry WS . 1998 Regulation of p53 downstream genes Semin. Cancer Biol. 8: 345–357
Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD . 1998 Activation of the ATM kinase by ionizing radiation and phosphorylation of p53 Science 281: 1677–1679
Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D . 1999 Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage Proc. Natl. Acad. Sci. USA 96: 14973–14977
Geyer RK, Yu ZK, Maki CG . 2000 The MDM2 RING-finger domain is required to promote p53 nuclear export Nature Cell Biol. 2: 569–573
Hengstermann A, Linares LK, Ciechanover A, Whitaker NJ, Scheffner M . 2001 Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells Proc. Natl. Acad. Sci. USA 98: 1218–1223
Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr Cj, Zambetti GP . 2000 p53-independent functions of the p19(ARF) tumor suppressor Genes Dev. 14: 2358–2365
Zhang Y, Xiong Y . 2001 A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation Science 292: 1910–1915
Shiloh Y . 2001 ATM and ATR: networking cellular responses to DNA damage Curr. Opin. Genet. Dev. 11: 71–77
Brown EJ, Baltimore D . 2000 ATR disruption leads to chromosomal fragmentation and early embryonic lethality Genes Dev. 14: 397–402
Gottrfredi V, Shieh SY, Taya Y, Prives C . 2001 p53 accumulates but is functionally impaired when DNA synthesis is blocked Proc. Natl. Acad. Sci. USA 98: 1036–1041
Takimoto R, El-Deiry WS . 2001 DNA replication blockade impairs p53-transactivation Proc. Natl. Acad. Sci. USA 98: 781–783
Chene P, Fuchs J, Bohn J, Garcia-Echeverria C, Furet P, Fabbro D . 2000 A small synthetic peptide, which inhibits the p53-hdm2 interaction, stimulates the p53 pathway in tumour cell lines J. Mol. Biol. 299: 245–253
Waterman MJF, Stavridi ES, Waterman JLF, Halazonetis TD . 1998 ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins Nature Genet. 19: 175–178
Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E . 1998 DNA damage activates p53 through a phosphorylation-acetylation cascade Genes Dev. 12: 2831–2841
Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y . 2000 p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by ser-46-phosphorylated p53 Cell 102: 849–862
Cho YJ, Gorina S, Jeffrey PD, Pavletich NP . 1994 Crystal structure of a p53 tumor-suppressor DNA complex – Understanding tumorigenic mutations Science 265: 346–355
Kaelin WG . 1999 The p53 gene family Oncogene 18: 7701–7705
Hibi K, Trink B, Patturajan M, Westra WH, Caballero OL, Hill DE, Ratovitski EA, Jen J, Sidransky D . 2000 AIS is an oncogene amplified in squamous cell carcinoma Proc. Natl. Acad. Sci. USA 97: 5462–5467
Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D . 1997 Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers Cell 90: 809–819
Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu WG, Flores ER, Tsai KY, Jacks T, Vousden KH, Kaelin . 2000 Role for the p53 homologue p73 in E2F-1-induced apoptosis Nature 407: 645–648
Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, Duyk G, Friedman L, Prives C, Kopczynski C . 2000 Drosophila p53 is a structural and functional homolog of the tumor suppressor p53 Cell 101: 91–101
Brodsky MH, Nortstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM . 2000 Drosophila p53 binds a damage response element at the reaper locus Cell 101: 103–113
Prabhu NS, Somasundaram K, Satyamoorthy K, Herlyn M, El-Deiry WS . 1998 p73 beta, unlike p53, suppresses growth and induces apoptosis of human papillomavirus E6-expressing cancer cells Int. J. Oncol. 13: 5–9
Agami R, Bernards R . 2000 Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage Cell 102: 55–66
Jimenez GS, Nister M, Stommel JM, Beeche M, Barcarse EA, Zhang XQ, O'Gorman S, Wahl GM . 2000 A transactivtaion-deficient mouse model provides insights into Trp53 regulation and function Nature Genet. 26: 37–43
Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW . 1999 Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition Science 284: 156–159
Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW . 2001 Inactivation of the apoptosis effector Apaf-1 in malignant melanoma Nature 409: 207–211
Zhao RB, Gish K, Murphy M, Yin YX, Notterman D, Hoffman WH, Tom E, Mack D, Levine AJ . 2000 Analysis of p53-regulated gene expression patterns using oligonucleotide arrays Genes Dev. 14: 981–993
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N . 2000 Noxa, a BH3-only member of the Bc1-2 family and candidate mediator of p53-induced apoptosis Science 288: 1053–1058
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B . 2001 PUMA induces rapid apoptosis of colorectal cancer cells Mol. Cell 7: 673–682
Nakano K, Vousden KH . 2001 PUMA, a novel proapoptotic gene, is induced by p53 Mol. Cell 7: 683–694
Lindsten T, Ross AJ, King A, Zong W-X, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB . 2000 The combined functions of proapoptotic Bc1-2 family members Bak and Bax are essential for normal development of multiple tissues Mol. Cell 6: 1389–1399
Lin YP, Ma WL, Benchimol S . 2000 Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis Nature Genet. 26: 122–125
Chinnaiyan AM, Prasad U, Shankar S, Hamstra DA, Shanaiah M, Chenevert TL, Ross BD, Rehemtulla A . 2000 Combined effect of tumor necrosis factor-related apoptosis-inducing ligand and ionizing radiation in breast cancer therapy Proc. Natl. Acad. Sci. USA 97: 1754–1759
Nagane M, Huang HJS, Cavenee WK . 2001 The potential of TRAIL for cancer chemotherapy Apoptosis 6: 191–197
Cuello M, Ettenberg SA, Nau MM, Lipkowitz S . 2001 Synergistic induction of apoptosis by the combination of TRAIL and chemotherapy in chemoresistant ovarian cancer cells Gynecol. Oncol. 81: 380–390
Mizutani Y, Nakao M, Ogawa O, Yoshida O, Bonavida B, Miki T . 2001 Enhanced sensitivity of bladder cancer cells to tumor necrosis factor related apoptosis inducing ligand mediated apoptosis by cisplatin and carboplatin J. Urol. 165: 263–270
Nagane M, Pan GH, Weddle JJ, Dixit VM, Cavanee WK, Huang HJS . 2000 Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo Cancer Res. 60: 847–853
Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S . 1999 Chemotherapy augments TRAIL-induced apoptosis in breast cell lines Cancer Res. 59: 734–741
Hengartner MO . 2000 The biochemistry of apoptosis Nature 407: 770–776
Ozoren N, Kim KH, Burns TF, Dicker DT, Moscioni AD, El-Deiry WS . 2000 The caspase 9 inhibitor Z-LEHD-FMK protects human liver cells while permitting death of cancer cells exposed to tumor necrosis factor-related apoptosis-inducing ligand Cancer Res. 60: 6259–6265
Burns TF, Bernhard EJ, El-Deiry WS . 2001 Tissue specific expression of p53 target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis in vivo Oncogene 20: 4601–4612
Fuchs EJ, McKenna KA, Bedi A . 1997 p53-dependent DNA damage-induced apoptosis requires Fas/APO-1-independent activation of CPP32 beta Cancer Res. 57: 2550–2554
Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ . 1995 Bax-deficient mice with lymphoid hyperplasia and male germ-cell death Science 270: 96–99
Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS . 1999 Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons J. Neurosci. 19: 7860–7869
Wei MC, Zong WX, Cheng EHY, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacCregor GR, Thompson CB, Korsmeyer SJ . 2001 Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death Science 292: 727–730
McDonald ER, Chui PC, Martelli PF, Dicker DT, El-Deiry WS . 2001 Death domain mutagenesis of KILLER/DR5 reveals residues critical for apoptotic signaling J. Biol. Chem. 276: 1439–1445
Merritt AJ, Poten CS, Kemp CJ, Hickman JA, Balmain A, Lane DP, Hall PA . 1994 The role of p53 in spontaneous and radiation-induced apoptosis in the gastrointestinal tract of normal and p53-deficient mice Cancer Res. 54: 614–617
Botchkarev VA, Komarova EA, Siebenhaar F, Botchkareva NV, Komarov PG, Maurer M, Gilchrest BA, Gudkov AV . 2000 p53 is essential for chemotherapy-induced hair loss Cancer Res. 60: 5002–5006
Wu GS, Burns TF, McDonald ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, El-Deiry WS . 1997 KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene Nature Genet. 17: 141–143
Kastan M . 1997 On the TRAIL from p53 to apoptosis? Nature Genet. 17: 130–131
Takimoto R, El-Deiry WS . 2000 Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site Oncogene 19: 1735–1743
Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS . 1999 Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor Cancer Res. 59: 2770–2775
Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, Blenis J, Tschopp T . 2000 TRAIL receptor-2 signals apoptosis through FADD and caspase-8 Nature Cell Biol. 2: 241–243
Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ . 2000 Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN Nature Med. 6: 529–535
Ozoren N, Fisher MJ, Kim K, Liu CX, Genin A, Shifman Y, Dicker DT, Spinner NB, Lisitsyn NA, El-Deiry WS . 2000 Homozygous deletion of the death receptor DR4 gene in a nasopharyngeal cancer cell line is associated with TRAIL resistance Int. J. Oncol. 16: 917–925
Pai SI, Wu GS, Ozoren N, Wu L, Jen J, Sidransky D, El-Deiry WS . 1998 Rare loss-of-function mutation of a death receptor gene in head and neck cancer Cancer Res. 58: 3513–3518
Lee SH, Shin MS, Kim HS, Lee HK, Park WS, Kim SY, Lee JH, Han SY, Park JY, Oh RR, Jang JJ, Han JY, Lee JY, Yoo NJ . 1999 Alterations of the DR5/TRAIL receptor 23 gene in non-small cell lung cancers Cancer Res. 59: 5683–5686
Shin MS, Kim HS, Lee SH, Park WS, Kim SY, Park JY, Lee JH, Lee SK, Lee SN, Jung SS, Han JY, Kim H, Lee JY, Yoo NJ . 2001 Mutations of tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1) and receptor 2 (TRAIL R2) genes in metastatic breast cancers Cancer Res. 61: 4942–4946
Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H, Okumura K . 2001 Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells Nature Med. 7: 94–100
Meng RD, El-Deiry WS . 2001 p53-Independent upregulation of KILLER/DR5 TRAIL receptor expression by glucocorticoids and interferon-gamma Exp. Cell Res. 262: 154–169
Fisher MJ, Virmani AK, Wu L, Aplenc R, Harper JC, Powell SM, Rebbeck TR, Sidransky D, Gazdar AF, El-Deiry WS . 2001 Nucleotide substitution in the ectodomain of TRAIL receptor DR4 is associated with lung cancer and head and neck cancer Clin. Cancer Res. 7: 1688–1697
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JCL, Lynch D . 1999 Tumoricidal activity of tumor necrosis factor related apoptosis-inducing ligand in vivo Nature Med. 5: 157–163
Jin Z, El-Deiry WS . 2001 Enhanced sensitivity of G1 arrested human cancer cells suggests a novel therapeutic strategy using a combination of Simvastatin and TRAIL Proc. Amer. Assoc. Cancer Res. 42: 438
Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR, Strom SC . 2000 Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand Nature Med. 6: 564–567
Nagata S . 2000 Steering anti-cancer drugs away from the TRAIL Nature Med. 6: 502–503
Lawrence D, Shakrokh Z, Marsters S, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, Hooley J, Sherwood S, Pai R, Leung S, Khan L, Gliniack B, Bussiere J, Smith CA, Strom SS, Kelley S, Fox JA, Thomas D, Ashkenazie A . 2001 Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions Nature Med. 7: 383–385
Takimoto R, El-Deiry WS . 2001 The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells, and can stabilize wild-type p53 protein Proc. Amer. Assoc. Cancer Res. 42: 922
Ricci MS, El-Deiry WS . 2000 Novel strategies for therapeutic design in molecular oncology using gene expression profiles Curr. Opin. Mol. Ther. 2: 682–690
Acknowledgements
WS El-Deiry is an Assistant Investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
El-Deiry, W. Insights into cancer therapeutic design based on p53 and TRAIL receptor signaling. Cell Death Differ 8, 1066–1075 (2001). https://doi.org/10.1038/sj.cdd.4400943
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400943
Keywords
This article is cited by
-
Exploiting loss of heterozygosity for allele-selective colorectal cancer chemotherapy
Nature Communications (2020)
-
Synergistic antitumor effect of AAV-mediated TRAIL expression combined with cisplatin on head and neck squamous cell carcinoma
BMC Cancer (2011)
-
Synergistic induction of cell death in liver tumor cells by TRAIL and chemotherapeutic drugs via the BH3-only proteins Bim and Bid
Cell Death & Disease (2010)
-
Treatment combinations targeting apoptosis to improve immunotherapy of melanoma
Cancer Immunology, Immunotherapy (2009)
-
Programmed Cell Death Pathways and Current Antitumor Targets
Pharmaceutical Research (2009)
