
Abstract
Neuronal cell death, abnormal protein aggregates, and cytoplasmic vacuolization are major pathologies observed in many neurodegenerative disorders such as the polyglutamine (polyQ) diseases, prion disease, Alzheimer disease, and the Lewy body diseases, suggesting common mechanisms underlying neurodegeneration. Here, we have identified VCP/p97, a member of the AAA+ family of ATPase proteins, as a polyQ-interacting protein in vitro and in vivo, and report on its characterization. Endogenous VCP co-localized with expanded polyQ (ex-polyQ) aggregates in cultured cells expressing ex-polyQ, with nuclear inclusions in Huntington disease patient brains, and with Lewy bodies in patient samples. Moreover, the expression of VCP mutants with mutations in the 2nd ATP binding domain created cytoplasmic vacuoles, followed by cell death. Very similar vacuoles were also induced by ex-polyQ expression or proteasome inhibitor treatment. These results suggest that VCP functions not only as a recognition factor for abnormally folded proteins but also as a pathological effector for several neurodegenerative phenotypes. VCP may thus be an ideal molecular target for the treatment of neurodegenerative disorders. Cell Death and Differentiation (2001) 8, 977–984
Similar content being viewed by others


Introduction
A class of inherited neurodegenerative disorders, including Huntington disease (HD) and Machado-Joseph Disease (MJD), is now known to be caused by expanded CAG repeats encoding for polyglutamines in the responsible genes.1,2 Expanded polyglutamines (ex-polyQs), typically of more than 40 repeats, have been shown to possess an intrinsic ability to aggregate in a polyQ length- and a polyQ concentration-dependent manner. The ability of polyQs to induce neurodegeneration in mice, and cell death in cultured cells appear to be inseparable from their intrinsic ability to aggregate, although there has been a report that the aggregates themselves are not essential for pathogenesis.3 This class of neurodegenerative disorders has thus been collectively called the ‘polyglutamine diseases’. Accordingly, nuclear and/or cytoplasmic ex-polyQ-containing aggregates or inclusions have been observed in neurons of patients suffering from essentially all the polyQ diseases. Accumulation of abnormal proteins has also been observed in various other human neurodegenerative disorders1 (e.g. prion disease, Alzheimer disease, amyotrophic lateral sclerosis, and the Lewy body diseases such as Parkinson disease and dementia with Lewy bodies). It is notable that cytoplasmic vacuolization is another hallmark which has long been observed in a broad range of neurodegenerations, as well as during aging;4 cytoplasmic vacuoles have been observed in the affected neurons of several polyQ diseases,5,6 Parkinson disease,7 prion disease,8 and Alzheimer disease.9 Very little is known however about these vacuoles at the molecular level.
Several experimental model systems have previously been developed for the molecular and genetical analyses of the accumulation of abnormal proteins, and for the analysis of its relationship to neurodegeneration.10,11,12 In yeast, the [PSI+] element is inherited by a prion-like mechanism involving self-propagating Sup35p aggregates.13 It has been shown that the propagation of not only Sup35p, but also that of polyglutamine aggregates depends on the quantity of the chaperon Hsp104p (heat shock protein 104); both an increase and a decrease of Hsp104p levels decreases the formation of both types of aggregates.14,15,16 Hsp104p has two ATPase domains, and belongs to a class of the AAA (ATPases associated with diverse cellular activities) ATPase protein family with a hexameric ring structure (AAA+ class), which also includes NSF (N-ethylmaleimide-sensitive fusion protein), VCP (valosin-containing protein)/p97/Cdc48p (cell division cycle protein 48), etc.17 A mammalian counterpart of Hsp104p however has not yet been identified. Here we report that VCP, a member of the AAA+ class of ATPases is an effector of several neurodegenerative phenotypes in ex-polyQ-expressing neuronal cells.
Results
During the search for molecules in mammalian cells which interact with ex-polyQs, all mammalian cultured cells examined were found to contain a protein with a molecular weight of approximately 100 kDa which interacted with the glutathione S-transferase-fused MJD protein containing a 79 polyglutamine repeat, GST-MJD7918 much more strongly than that fused with the MJD protein containing a 35 polyglutamine repeat, GST-MJD35.18 This protein was then purified from both COS cells and HeLa cells by GST-MJD79-mediated affinity purification. Mass spectrometry and microsequencing analyses revealed that the protein from both cell types was VCP, a member of the AAA+ class of ATPases. Northern blot analyses of human RNA showed that VCP was expressed ubiquitously in all tissues (not shown) and throughout the brain (Figure 1A), suggesting the possibility that VCP could be involved in human neurodegenerations occurring in all brain areas.

Identification of VCP as a protein interacting with the MJD protein containing an expanded polyglutamine repeat. (A) VCP and control GAPDH mRNA expression in 16 different areas of the human brain was analyzed by Northern blot analysis on a membrane preblotted by Clontech. (B) In vitro synthesized 35S-labeled VCP shows stronger interaction with GST-MJD79 than with GST-MJD35, and no interaction with GST. (C) Identification of the domain of MJD79 essential for its interaction with VCP via the GST pull-down assay. The illustrations show schematic structures of the MJD79 deletions. The polyglutamine stretches are indicates as black boxes. (D) Identification of the domain of VCP essential for its interaction with MJD79 via the GST pull-down assay. The illustrations show schematic structures of VCP deletions. The Walker A and Walker B motifs of the ATP binding domains in VCP are indicated as black and gray boxes, respectively. The crosshatched areas highlight the domain of VCP needed for its interaction with MJD79. Autoradiographs show 20% of the total amount of 35S-labeled proteins used for the GST pull-down assay (input) and the pulled down proteins (GST-VCP, GST-MJD79). CBB staining of the GST-fused proteins used are also presented (CBB)
In vitro pull-down assays were performed in order to examine whether cloned VCP binds to MJD79. VCP indeed bound strongly to MJD79 and weakly to MJD35 (Figure 1B). Deletion analyses of MJD79 indicated that VCP binds directly to the stretch of ex-polyQs in the MJD protein (Figure 1C). Deletion analyses of VCP revealed that the portion containing amino acid residues 143–199, located towards the N-terminus, is responsible for its binding to MJD79 (Figure 1D). We have tried several different experiments to test whether VCP acts as a chaperon towards the ex-polyQs, but have not been able to reach any convincing conclusions as yet. We have however observed that like Hsp104p,19 VCP can also bind to Hsp70 and Hsp40 (data not shown).
Consistent with the in vitro binding data shown above, endogenous VCP was found to colocalize with ex-polyQ stretches. Overexpressed flag-tagged 79 polyglutamine repeat fragments, Q79, were found to exist as aggregates both in the nucleus and in the cytoplasm in neuronally differentiated PC12 cells,20 and to colocalize with endogenous VCP (Figure 2A,B). In contrast, normal length polyQs such as Q14 and Q26 showed little binding to VCP (not shown), and remained in the cytoplasm without forming aggregates when expressed in neuronal PC12 cells.20 Most of the Q79-expressing cells were found to contain cytoplasmic vacuoles (Figure 2B). In brain sections from HD and MJD, VCP appeared to exist in the nuclear inclusions (Figure 2C, and not shown). Furthermore, VCP-positive staining was observed in Lewy bodies (Figure 2D). The accumulation of VCP not only in the nuclear inclusions observed in the polyglutamine diseases but also in Lewy bodies indicate that VCP can recognize a broad range of abnormally folded proteins.

Co-localization of endogenous VCP with polyglutamine aggregates in PC12-Q79 cells, with nuclear inclusions in Huntington disease (HD), and with Lewy bodies in dementia with Lewy bodies (DLB). Endogenous VCP protein in PC12 cells (A) and PC12-Q79 cells (B) was stained with an anti-VCP antibody and visualized by FITC signals. Endogenous VCP was detected throughout the PC12 cells (A). Expressed Q79 was stained with an anti-flag antibody and visualized by Texas Red signals in PC12-Q79 cells, 48 h (upper panels) and 96 h (lower panels) after the induction of Q79. The yellow colors in the superimposed panels show the co-localization of endogenous VCP and Q79 (B). (C) Identification of endogenous VCP protein in nuclear inclusion-positive neurons from HD patients (arrows). Brain sections containing ubiquitin-positive nuclear inclusions from the postmortem cerebral cortex of Japanese patients with Huntington disease20 were stained with an anti-VCP antibody, and signals were visualized by the ABC method20 (brown signals). (D) Identification of endogenous VCP protein in Lewy bodies from DLB patients (an arrow). Brain sections containing Lewy bodies from the postmortem brains of patients with DLB were stained with an anti-VCP antibody, and signals were visualized by the ABC method20 (brown signals). Bars represent 20 μm (C, D)
A series of VCP point mutants were created covering the different portions to test the possibility of generating mutants which may mimic ex-polyQ-induced phenotypes in cultured cells. Among the many mutants (e.g. K251A, E305Q, L306P, Y495A, Y495E, K524A, E578Q, L579P, Y805A, and Y805E), only the K524A mutant with a substitution in the Walker A motif of the 2nd ATP binding domain clearly induced large cytoplasmic vacuoles in cells transfected with this mutant (Figure 3A). A deletion mutant of this Walker A motif (Δ517–525) also formed large vacuoles when transfected into cells (data not shown). Furthermore, cells transfected with VCP(K524A) and VCP(Δ517–525) eventually died following vacuole formation. NSF(K549A), a NSF mutant containing an equivalent mutation to VCP(K524A), has been reported to lose its ATPase activity and ATP binding, and has been proposed to function in a dominant-negative manner within the NSF hexameric complex.21 The predicted structure of NSF indicates that the N-terminal portion is juxtaposed to the second ATPase domain.22,23 The binding of certain abnormally folded proteins to the N-terminal portion of VCP may thus affect its ATPase activity and function similarly to VCP(K524A) and VCP(Δ517–525). NSF(K549A) however, was not able to induce such vacuoles or cell death (data not shown). Both vacuole formation and cell death induced by VCP(K524A) was suppressed by the co-expression of wild-type VCP in a dose-dependent manner (Figure 3B,C).

Vacuole formation caused by VCP(K524A) and its rescue by the expression of wild-type VCP. (A) Cytoplasmic vacuoles were induced by GFP-fused VCP(K524A) (right two panels) but not by GFP-fused wild-type VCP (left two panels) in COS, HeLa, and PC12 cells. (B, C) PC12 cells were co-transfected with VCP(K524A) in different ratios to wild-type VCP, and the percentages of cells containing vacuoles (B) and an apoptotic morphology (C) were examined 36 h after transfection
We previously observed that proteasome inhibitors such as MG115 and PSI caused the creation of numerous vacuoles in neuronal PC12 cells and HeLA cells, similar to those observed in VCP(K524A)-expressing cells (Figure 4A). The proteasome is a complex that mediates the degradation of unnecessary proteins within cells, misfolded proteins being the obvious target. Treatment of cells with proteasome inhibitors has been shown to raise the amount of misfolded proteins within cells,24 which is similar to the situation in degenerating neurons containing abnormally folded proteins. Indeed, PC12 cells treated with such proteasome inhibitors showed the accumulation of abnormal protein aggregates, and these aggregates were clearly stained with the anti-VCP antibody (Figure 4A). Overexpression of VCP suppressed the formation of vacuoles created by such proteasome inhibitors transiently, but not permanently (Figure 4B,C).

Proteasome treatment induces VCP-positive protein aggregates and large cytoplasmic vacuoles in PC12 cells. (A) Phase contrast views (upper panels) and immunohistochemical views (lower panels) of PC12 cells treated with 0.1 μM PSI for 48 h. Endogenous VCP protein was stained with an anti-VCP antibody and visualized by Texas Red signals (lower panels). Arrows indicate the protein aggregates observed in cells treated with proteasome inhibitors (upper panels). (B) Vacuole formation induced by 10 μM MG115 was suppressed by the expression of wild-type VCP. (C) The percentages of cells containing vacuoles were examined in the control or wild-type VCP transfected PC12 cells 24 h after treatment with 10 μM MG115 or 1 μM PSI. Transfected cells were identified with GFP signals (GFP)
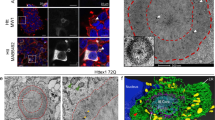
We analyzed cells expressing ex-polyQ or VCP(K524A) by electron microscopy. Both types of cells contained many morphologically similar vacuoles. Typical vacuoles observed in both types of cells had ribosome-positive membranes (Figure 5A,D). Considering the evidence that VCP is involved in vesicle trafficking from the endoplasmic reticulum (ER) to the Golgi, these vacuoles are likely to be a result of abnormal budding and enlargement of the ER. Some vacuoles appeared to be connected to the tubular ER (Figure 5B). Note that very few ribosomes were observed on the membranes of greatly enlarged vacuoles observed in the late states of ex-polyQ-expressing cells (Figure 5B). Detailed morphological analyses of the processes of these vacuole formations will be reported elsewhere. The accumulation of enlarged ER-like vacuoles morphologically similar to those observed in cells expressing VCP(K524A) have been observed in the motor neurons of ALS model mice expressing mutant SODs.25 These results strongly indicate that VCP itself is involved in vacuole formation in cells containing ex-polyQs and other misfolded proteins as well. VCP thus should be re-abbreviated for ‘vacuole-creating protein’.

Ultrastructural analyses of the vacuoles induced by polyglutamines and VCP(K524A). (A–D) Electron micrograph of PC12 cells expressing Q79 (A, B) and VCP(K524A) (C,D) under the control of the tet-off promoter. Cells were examined 24 h (A) and 48 h (B–D) after removal of tetracycline. Bars represent 1 μm (A,B,D) and 10 μm (C); arrows, membrane portions with ribosomes; arrowheads, fused portions between ER and abnormally enlarged vacuoles. The area shown in (D) is an enlarged view of the rectangle in (C)
Discussion
In this study, we have identified VCP, a member of the AAA+ class of ATPases, as an MJD79-interacting protein by a biochemical approach. We have also identified VCP as a modifier of polyglutamine-mediated eye degeneration in Drosophila via genetic screening. Our genetic study using Drosophila identified ter94/VCP as a factor that worsens ex-polyQ-induced eye degeneration, indicating that VCP functions as an effector (manuscript in preparation). Consistently, in this present study, several lines of evidence demonstrate that VCP functions as an effector for several neurodegenerative phenotypes observed in cultured polyQ-expressing neuronal cells, namely vacuole creation and cell death.
Various similarities can be noted regarding structure-function relationships within the AAA+ class of ATPases. The N-terminal portion of NSF is involved in protein binding, and binds to SNAP (soluble NSF attachment protein) via its N-terminus.26 Likewise, the portion of VCP close to its N-terminus is indispensable for its interaction with MJD79. Hsp104p has been reported to interact with Sup35p as well as mammalian Prsc, and Aβ, although the precise domains of interaction have not been identified.27 We found that VCP was also able to bind to an intracellular portion of APP (amyloid precursor protein) containing Aβ, as well as to Hsp40 and Hsp70. These observations consistently support the idea that Hsp104p and VCP have the intrinsic ability to act as a sensor of abnormally folded proteins, probably using portions near their N-terminus to sense misfolded proteins either directly or indirectly via other proteins such as Hsp40 and Hsp70.
A hexameric structure such as that of VCP has the theoretical advantage to function as a system for monitoring the concentration of target molecules in cells; it should be able to recognize at least six different states by monitoring how many binding sites among the six are occupied by target molecules. Following this hypothetical step, Hsp104p functions as a chaperon to the misfolded proteins, whereas VCP appears to perform a toxic function similar to that induced by mutations in its second ATP binding domain, leading to vacuole formation and eventually cell death. It is easily imaginable that such a toxic stoichiometry is increased in a certain age-dependent frequency in neurons of patients suffering from polyglutamine diseases as well as other neurodegenerations with abnormally folded proteins, or even in aged brains.4 If so, the invention of small molecules which inhibit binding between VCP and abnormally folded proteins will be another strategy for the treatment or prevention of many types of neurodegenerations, as well as brain aging.
Materials and Methods
GST pull-down assay
A series of 35S-labeled VCP or MJD proteins were prepared by the in vitro transcription and translation system (Promega). 35S-labeled proteins were mixed with the GST-fusion proteins in NETN buffer with protease inhibitors (100 mM NaCl, 1 mM EDTA, 20 mM Tris (pH 7.4), 0.5% NP-40, 100 μg ml−1 aprotinin, 100 μg ml−1 leupeptin, 1 mM PMSF) at 4°C with rotation for 2 h, followed by centrifugation and washing with buffer H (20 mM HEPES (pH 7.7), 50 mM KCl, 20% glycerol, 0.1% NP-40) repeated three times. Bound proteins were eluted from the beads with SDS sample buffer, boiled for 5 min, resolved by SDS–PAGE, and detected by autoradiography.
Cell lines, transfection and GFP assays
Establishment and maintenance of PC12-Q79 cells that express Q79, a flag-tagged 79 repeat of polyglutamine from the MJD protein, under the control of the tet-off promoter has been described previously.20 For GFP assays, transfection was performed using Transfast (Promega); 5×104 cells in a 6-well dish were transfected with 1 μg pCAGGS vector containing cDNAs together with 0.2 μg pEGFP vector (Clontech). Twenty-four hours after transfection, tetracycline was removed from the medium, and an additional 48, 72 or 96 h later, cell morphology and GFP signals were analyzed using a microscope. NGF was added to PC12 or PC12-Q79 cells at a concentration of 50 ng ml−1 at the time of tetracycline removal. The ratio of cells containing vacuoles and apoptotic features were determined by dividing the number of GFP-positive cells containing vacuoles or showing apoptotic morphologies (e.g. membrane blebbing and cell shrinkage) by the total number of GFP-positive cells (approximately 200 cells). In the Figures, the mean values of triplicate experiments are presented, and s.d. are included as bars. Statistic values from the Student t-test are also included; *P<0.05; **P<0.01.
VCP antibodies
The GST-VCP protein was expressed in E. Coli and thrombin-cleaved VCP protein was purified by FPLC (Pharmacia). Rabbits were immunized with the purified VCP protein following standard procedures. The resulting immune serum was affinity-purified against VCP proteins immobilized on AminoLink column (PIERCE).
Cell treatment for immunocytochemistry
Cells were rinsed with PBS and fixed with 4% paraformaldehyde or 100% ethanol for 10 min at room temperature. After fixation, cells were permeabilized with 0.2% Triton X-100 for 3 min and incubated at 4°C overnight with M5, a mouse monoclonal anti-flag antibody (Kodak), and/or a rabbit polyclonal anti-VCP antibody. Subsequently, cells were treated with Texas-Red conjugated and/or FITC conjugated secondary antibodies (Jackson Immuno Research) and mounted in anti-fade solution with DAPI (Vector Laboratories).
References
Kakizuka A . 1998 Protein precipitation: a common etiology in neurodegenerative disorders? Trends Genet. 14: 396–402
Price DL . 1999 New order from neurological disorders Nature 399: A3–5
Saudou F, Finkbeiner S, Devys D, Greenberg ME . 1998 Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions Cell 95: 55–66
Okamoto K, Hirai S, Iizuka T, Yanagisawa T, Watanabe M . 1991 Reexamination of granulovacuolar degeneration Acta Neuropathol. 82: 340–345
Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M . 1997 Huntingtin localization in brains of normal and Huntington's disease patients Ann. Neurol. 42: 604–612
Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT . 1998 Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice Cell 95: 41–53
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y . 1997 Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease Histol. Histopathol. 12: 25–31
Grigoriev V, Escaig-Haye F, Streichenberger N, Kopp N, Langeveld J, Brown P, Fournier JG . 1999 Submicroscopic immunodetection of PrP in the brain of a patient with a new-variant of Creutzfeldt-Jakob disease Neurosci. Lett. 264: 57–60
Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA . 1996 Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease J. Neurosci. 16: 186–199
Kakizuka A . 1997 Degenerative ataxias: genetics, pathogenesis and animal models Curr. Opin. Neurol. 10: 285–290
Bates GP, Mangiarini L, Davies SW . 1998 Transgenic mice in the study of polyglutamine repeat expansion diseases Brain Pathol. 8: 699–714
Fortini ME, Bonini NM . 2000 Modeling human neurodegenerative diseases in Drosophila: on a wing and a prayer Trends Genet. 16: 161–167
Kushnirov VV, Ter-Avanesyan MD . 1998 Structure and replication of yeast prions Cell 94: 13–16
Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW . 1995 Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science 268: 880–884
Krobitsch S, Lindquist S . 2000 Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins Proc. Natl. Acad. Sci. USA 97: 1589–1594
Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI . 2000 Polyglutamine aggregates after protein folding homeostasis in Caenorhabditis elegans Proc. Natl. Acad. Sci. USA 97: 5750–5755
Neuwald AF, Aravind L, Spouge JL, Koonin EV . 1999 AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes Genome Res. 9: 27–43
Ikeda H, Yamaguchi M, Sugai S, Aze Y, Narumiya S, Kakizuka A . 1996 Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo Nat. Genet. 13: 196–202
Glover JR, Lindquist S . 1998 Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins Cell 94: 73–82
Yasuda S, Inoue K, Hirabayashi M, Higashiyama H, Yamamoto Y, Fuyuhiro H, Komure O, Tanaka F, Sobue G, Tsuchiya K, Hamada K, Sasaki H, Takeda K, Ichijo H, Kakizuka A . 1999 Triggering of neuronal cell death by accumulation of activated SEK1 on nuclear polyglutamine aggregations in PML bodies Genes Cells 4: 743–756
Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE . 1994 N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion J. Cell Biol. 126: 945–954
Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE . 1997 Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy Cell 90: 523–535
May AP, Misura KM, Whiteheart SW, Weis WI . 1999 Crystal structure of the amino-terminal domain of N-ethylmaleimide-sensitive fusion protein Nat. Cell Biol. 1: 175–182
Bush KT, Goldberg AL, Nigam SK . 1997 Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance J. Biol. Chem. 272: 9086–9092
Dal Canto MC, Gurney ME . 1995 Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS) Brain Res. 676: 25–40
Nagiec EE, Bernstein A, Whiteheart SW . 1995 Each domain of the N-ethylmaleimide-sensitive fusion protein contributes to its transport activity J. Biol. Chem. 270: 29182–29188
Schirmer EC, Lindquist S . 1997 Interactions of the chaperone Hsp104 with yeast Sup35 and mammalian PrP Proc. Natl. Acad. Sci. USA 94: 13932–13937
Acknowledgements
We thank H Yoshii and M Sugimoto for secretarial assistance in the preparation of this manuscript. We thank G Sobue (Nagoya University) for postmortem brain samples with HD, and M Baba and T Iwatsubo (Tokyo University) for postmortem brain samples for DLB. This work was supported in part by research grants from the Ministry of Education, Science, Sports, and Culture of Japan, and the Ministry of Welfare of Japan, the Yamanouchi Foundation for Research on Metabolic Disorders, the Naito Foundation, and the Uehara Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by H Ichijo
Rights and permissions
About this article
Cite this article
Hirabayashi, M., Inoue, K., Tanaka, K. et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ 8, 977–984 (2001). https://doi.org/10.1038/sj.cdd.4400907
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400907
Keywords
This article is cited by
-
Autophagy Dysfunction in ALS: from Transport to Protein Degradation
Journal of Molecular Neuroscience (2022)
-
VCP/p97 regulates Beclin-1-dependent autophagy initiation
Nature Chemical Biology (2021)
-
Gossypol, a novel modulator of VCP, induces autophagic degradation of mutant huntingtin by promoting the formation of VCP/p97-LC3-mHTT complex
Acta Pharmacologica Sinica (2021)
-
Synergistic killing effect of paclitaxel and honokiol in non-small cell lung cancer cells through paraptosis induction
Cellular Oncology (2021)
-
A Brazilian family with inclusion body myopathy associated with Paget’s disease of bone and frontotemporal dementia linked to the VCP pGly97Glu mutation
Clinical Rheumatology (2018)
