
Abstract
The ubiquitin-proteasome pathway plays a critical role in the degradation of several proteins involved in the cell cycle. Dysregulation of this pathway leads to inhibition of cellular proliferation and the induction of apoptosis. Ubiquitination and its downstream consequences have been investigated intensively as targets for the development of drugs for tumour therapy. Here we have investigated the mechanism of apoptosis induced by the proteasome inhibitors MG-132, lactacystin and calpain inhibitor I (ALLN), in the HEK 293 cell line and the ovarian cancer cell lines SKOV3 and OVCAR3. We have found strong caspase-3-like and caspase-6-like activation upon treatment of HEK 293 cells with MG-132. Using a tricistronic expression vector based on a tetracycline-responsive system we generated stable SKOV3 nd OVCAR3 cell lines with inducible expression of pro-caspase-3. Induction of pro-caspase-3 expression in normally growing cells does not induce apoptosis. However, in the presence of the proteasome inhibitors MG-132, lactacystin or ALLN we found that cells overexpressing pro-caspase-3 are rapidly targeted for apoptosis. Our results demonstrate that pro-caspase-3 can sensitise ovarian cancer cells to proteasome inhibitor-induced apoptosis, and a combination of these approaches might be exploited for therapy of ovarian and other cancers. Cell Death and Differentiation (2001) 8, 256–264
Similar content being viewed by others


Introduction
Apoptosis is a form of cell death involved in the regulation of a wide range of physiological systems including normal cell turnover, control of the immune system, embryonic development and hormone-dependent tissue atrophy. Inappropriate apoptosis has been implicated in diseases ranging from Alzheimer's disease, autoimmune disorders, to AIDS and cancer. Many studies have shown that signalling through so-called ‘survival’ pathways not only provides stimulation for proliferation but can also block cell death pathways. Growth factors signalling through the PI3 kinase/Akt pathway promote cell survival by inhibiting apoptosis and the processing of procaspases to their active forms.1,2,3 It is also evident that treatment with various anticancer drugs leads to selection of tumour cells more resistant to apoptosis.4,5,6,7
Caspases are a large family of cysteine proteases, most of them playing central roles in the execution of apoptosis.8 Two main pathways involved in the activation of caspases have been described so far. The first is initiated by the death factors, leading to the activation of caspase-8 by its recruitment to the death receptor. Activated caspase-8 can activate downstream effector caspases, such as caspases-3, -6 and -7.9,10,11 The second pathway is initiated by the release of cytochrome c from mitochondria. This leads to activation of caspase-9 by formation of a complex with Apaf-1/cytochrome c. Activated caspase-9 will then cleave and activate the downstream effector caspases-3, -6 and -7.12,13,14 Once activated by either pathway, effector caspases can disassemble the cell by cleavage of the vast majority of key cellular proteins.15
Interestingly, overexpression of effector caspases in mammalian cells is relatively non-toxic and this makes them potential tools for gene therapy of cancer in combination with caspase-activating drugs. It has been shown that the combination of pro-caspase-3 overexpression with exposure to etoposide can enhance apoptosis in an AH130 liver tumour model.16 Similar results have been found upon treatment of U-373MG glioma cells overexpressing pro-caspase-3 with Fas ligand.17
Several reports have shown a tight connection between apoptosis and the activity of the ubiquitin-proteasome pathway. In human leukaemia HL60 cells, inhibition of proteasome-mediated proteolysis by specific proteasome inhibitors led to the rapid induction of apoptosis.18 Calpain inhibitor I can induce p53-dependent apoptosis in MOLT-4 and L51784 cells.19 Blocking proteasome function by inhibitors such as MG-132 or lactacystin leads to induction of apoptosis with activation of caspase-3 and Bcl-2 cleavage.20 Recently, a new series of proteasome inhibitors has been tested for their potential in cancer therapy. By blocking protein turnover, these agents caused inhibition of tumour growth as well as the induction of apoptosis.21
In this study we have investigated the effect of different proteasome inhibitors on ovarian cancer cell lines overexpressing pro-caspase-3. By exploiting a new tri-cistronic, tet-inducible vector we have found that overexpression of pro-caspase-3 alone is not itself sufficient to induce apoptosis but can dramatically sensitise cells to the effect of exposure to proteasome inhibitors.
Results
Proteaome inhibitors induce G2/M cell cycle arrest and caspase-3- and caspase-6-like activity but not caspase-1-like activity in HEK 293 cells
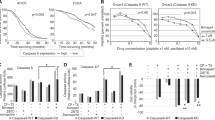
Proteasome inhibitors can induce apoptosis by blocking protein turnover in the cell. The main targets of proteasomes are various classes of cell cycle regulatory proteins and dysregulation of their levels would be expected to affect the cell cycle. To test how proteasome inhibitors affect the cell cycle, HEK 293 cells were incubated with MG-132 for 24 h and analyzed immediately or transferred to medium without the inhibitor and incubated in normal conditions for another 24 h. The cell cycle status of the cells was analyzed with a FACSCalibur flow cytometer by measuring fluorescence from cells stained with propidium iodide. After 24 h incubation with MG-132, the percentage of cells in G1 phase was decreased and the percentage of cells in G2/M was increased (Figure 1B). After further incubation for 24 h in normal medium the cells showed arrest in G2/M-phase and appearance of apoptotic cells in the sub-G1 region (Figure 1C).

Flow cytometric analysis of HEK 293 cells treated with MG-132 for different times. DNA was stained with propidium iodide and DNA content determined by flow cytometry. Data are presented as percentage subG1 region (M1), G1 phase (M2), S-phase (M3) and G2/M-phase (M4). (A) Control HEK 293 were grown in DMEM-10% FCS and the cell cycle status was analyzed by measuring fluorescence from cells stained with propidium iodide. (B) HEK 293 cells were grown in the presence of 10 μM MG-132 for 24 h and the cell cycle status was analyzed as in (A). (C) HEK 293 cells were grown in the presence of 10 μM MG-132 for 24 h and thereafter in normal medium for another 24 h; cell cycle status was analyzed as in (A)
To check whether caspase activity is involved in the MG-132 induced apoptosis we treated 293 cells with different concentrations of inhibitor and after 48 h cells were lysed and caspase-3- and 6-like activities was measured against fluorogenic substrates Ac-DEVD-AMC (Figure 2A) and Ac-VEID-AFC (Figure 2B) respectively. The effect of MG-132 was compared with taxol, an apoptosis-inducing drug which is used clinically for the treatment of ovarian and other forms of cancer.22,23 Prominent caspase-3- and -6-like activities were observed when the cells were treated with MG-132. In the same time taxol was not able to induce significant caspase-3- and -6-like activities.

Proteasome inhibitor MG-132 induces caspase-3-like and caspase-6-like activation. HEK 293 cells were incubated with various concentrations of MG-132 or taxol for 48 h and lysed in caspase assay lysis buffer (see Materials and Methods). Caspase-3-like (A) and caspase-6-like (B) activities were measured against fluorometric substrates Ac-DEVD-AMC and Ac-VEID-AFC respectively (see Materials and Methods)
In order to prove involvement of caspase-3 in apoptosis induced by the proteasome inhibitor we transiently overexpressed pro-caspase-3 in 293 cells and treated the cells with the proteasome inhibitor MG-132. After 24 h, treated and control cells were lysed and caspase-1-, -3 and -6-like activities were measured against the corresponding fluorogenic substrates (Figure 3). Caspase-1 is not involved in apoptosis and its activity was not affected by overexpression of pro-caspase-3 alone or in combination with the proteasome inhibitor MG-132. Overexpression of pro-caspase-3 alone resulted in an increase in its own activity without altering the activity of the downstream caspase-6 (Figure 3), which indicates that this intervention is not enough to induce activation of other caspases. However combination of pro-caspase-3 overexpression with the proteasome inhibitor MG-132 showed very strong caspase-3- and -6-like activation which clearly demonstrates involvement of both caspases in MG-132-induced apoptosis (Figure 3).

Pro-caspase-3 overexpression in HEK 293 cells enhances caspase-3-like and caspase-6-like activaties against proteasome inhibitor MG-132. HEK 293 cells were transiently transfected with control vector (pcDNA3) or pro-caspase-3-expressing vector (pcDNA3pro-caspase-3) by calcium phosphate coprecipitation. After 24 h cells were incubated in the presence or absence of 10 μM MG-132 for another 24 h and lysed in caspase assay lysis buffer (see Materials and Methods). Caspase-1-, -3- and -6-like activities were measured against fluorometric substrates Ac-YVAD-AMC, Ac-DEVD-AMC and Ac-VEID-AFC respectively (see Materials and Methods)
Pro-caspase-3 overexpression alone does not induce apoptosis in HEK 293 cells
To test whether enhanced caspase-3-like activity as a result of overexpression in 293 cells is alone enough to induce apoptosis we analyzed cell morphology. Although the level of pro-caspase-3 expression was several fold more than the endogenous level there was no significant difference in cellular morphology when we compared pro-caspase-3 transfected 293 cells with control cells tranfected with empty vector (Figure 4A,C) However, treatment of the cells with MG-132 for 48 h or transfection of constitutively active caspase-3 showed clear induction of apoptosis which was blocked by caspase inhibitor-Ac-DEVD-CHO (Figure 4B,D,E). These results were additionally supported by analysis of PARP cleavage, which showed no change in cells overexpressing pro-caspase-3 (data not shown). These results demonstrate that pro-caspase-3 alone is not enough to induce apoptosis in HEK 293 cells.

Pro-caspase-3 overexpression does not induce apoptosis. HEK 293 cells were transiently transfected with pcDNA3 expression plasmids encoding control vector, pro-caspase-3 andd reverse caspase-3 or incubated with 10 μM MG-132. Phase contrast microscopy of HEK 293 cells at 48 h after transfection or exposure to the inhibitor. (A) Control vector. (B) MG-132 treatment. (C) Pro-caspase-3 expression vector. (D, E) Reverse caspase-3 (constitutively active form) expression vector in absence (D) or presence (E) of 100 μM caspase inhibitor DEVD-CHO
Generation of SKOV3 and OVCAR3 stable cell lines expressing pro-caspase-3 under inducible control
To generate stable cell lines expressing pro-caspase-3 from ovarian cancer cell lines SKOV3 and OVCAR3 a tricistronic, tetracycline-responsive expression vector pNRTIS-33 was used.24 Stable derivatives of the OVCAR3 and SKOV3 cell lines were generated after transfection with control or pro-caspase-3 expression vectors. Several clones inducibly expressing pro-caspase 3 were isolated. The level of pro-caspase-3 expression was suppressed in the presence of ATc (anhydrotetracycline) and induced in its absence (Figure 5, lanes 3 and 4). The results from inducible expression of pro-caspase-3 confirmed that an increased level of this caspase is not sufficient to induce apoptosis.

Generation of stable cell lines expressing inducible pro-caspase-3. SKOV3 cells were transfected with tricistronic, tetracycline-regulatable vector (pNRTIS-33) or pNRTIS pro-caspase-3 using Superfect transfection reagent. Several clones were isolated by selection in medium containing 1 mg/ml G-418 and subsequently incubated in the presence or absence of 100 ng/ml ATc for 72 h. Cell lysates were subjected to immunoblotting with anti-caspase-3 antibody (Santa Cruz). Lanes 1 and 2, clone SKK-1 transfected with control vector; lanes 3 and 4, clone SK-1 transfected with pNRTIS-33 pro-caspase-3
Pro-caspase-3 overexpression sensitises SKOV3 and OVCAR3 cell lines to apoptosis induced by proteasome inhibitors
To test the effect of proteasome inhibitors on SKOV3 stable cell lines (SKK-1, transfected with control vector and SK-1, transfected with pro-caspase-3) we treated the cells with three different proteasome inhibitors MG-132 (15 μM), ALLN (100 μM) and lactacystin (12.5 μM). Lactacystin is a specific inhibitor of the 20S proteasome, while MG-132 and ALLN are potent but less specific proteasome inhibitors. As a control we included taxol. Cells were lysed at different time points and caspase-3-like activity was measured against the colorimetric substrate Ac-DEVD-pNA (see Materials and Methods). After 12 h treatment with each of the proteasome inhibitors we found very strong caspase-3-like activation in SK-1 cells which had induced pro-caspase-3 expression (Figure 6, lanes 15, 17 and 19). In cells transfected with the pro-caspase-3 expression construct in which transcription of the transgene was suppressed there was reduced activity (Figure 6, lanes 16, 18 and 20), but still higher than in the control cell line (Figure 6, lanes 4–9).These results can be explained by the slight leakage of pro-caspase-3 expression after suppression with ATc which leads to a slightly higher pro-caspase-3 level than the control cell line (Figure 5, compare lanes 1 and 3). The effect of taxol on caspase-3-like activation was not as marked as observed for the proteasome inhibitors but exposure to this drug still induced some sensitisation (Figure 6, lanes 21 and 22). Caspase-3 activity was also characterised by analyzing PARP cleavage and the presence of active caspase-3. After 12 h of treatment with MG-132 we were able to see the 85 kDa PARP cleavage product and the appearance of the active form of pro-caspase-3 only in the SK-1 cells where pro-caspase-3 was induced (Figure 7, lanes 8 and 9). There was no PARP cleavage detected in the control cell line (Figure 7, lanes 2, 3, 5 and 6) which indicates that apoptosis induced by the proteasome inhibitor is dependent on the level of pro-caspase-3. Characterisation of cell morphology by phase-contrast microscopy supported these results. As shown in Figure 8, treatment of the control clone SKK-1 with 10 μM MG-132 induced the appearance of only a few apoptotic cells (Figure 8E,F) compared to the non-treated cells (Figure 8A,B). In contrast, massive apoptosis was observed in the pro-caspase-3 overexpressing clone SK-1 on treatment with MG-132 (Figure 8G). Suppression of pro-caspase-3 expression with ATc reduced the level of apoptosis induced with MG-132 (Figure 8H). These results confirm that overexpression of pro-caspase-3 alone cannot induce apoptosis but its level of expression is a crucial factor for apoptosis induced with MG-132.

Effect of different proteasome inhibitors and taxol on caspase-3-like activity in SKOV3 stable transfectants. Clone SKK-1 (control-transfected) and clone SK-1 (pNRTIS-33 pro-caspase-3) were grown in the absence or presence (100 ng/ml) of ATc for 5 days and were incubated with various proteasome inhibitors and taxol as indicated. Cells were lysed and caspase-3-like activity was measured against the colorimetric substrate-Ac-DEVD-pNA. Lane 1, substrate alone; lanes 2–22 plus cytoplasmic extracts from cells treated as indicated

Effect of MG-132 on PARP cleavage in SKOV3 stable transfectants. Clone SKK-1 (control-transfected) and clone SK-1 (pNRTIS-33 pro-caspase-3) were grown in the absence or presence (100 ng/ml) of ATc for 5 days and were incubated in the absence or presence (10 μM) of MG-132, as indicated. Cells were lysed in Triton buffer and lysates were subjected to immunoblotting with anti-PARP antibody (upper panel) or anti-active caspase-3 antibody (lower panel)

Morphology of MG-132 treated SKOV3 cells with or without expression of pro-caspase-3. SKOV3 stable cell lines SKK-1 (control) and SK-1 (pro-caspase-3) were grown for 5 days in presence or absence of 100 ng/ml ATc. Cells were then treated with or without 10 μM MG-132 and were observed under a phase-contrast microscope after 12 h. (A) Untreated control clone SKK-1 without ATc. (B) Untreated control clone SKK-1 with 100 ng/ml ATc. (C) Untreated pro-caspase-3 stable clone SK-1 without ATc. (D) Untreated pro-caspase-3 stable clone SK-1 with 100 ng/ml ATc. (E) MG-132 treated control clone SKK-1 without ATc. (F) MG-132 treated control clone SKK-1 with 100 ng/ml ATc. (G) MG-132 treated pro-caspase-3 stable clone SK-1 without ATc. (H) MG-132 treated pro-caspase-3 stable clone SK-1 with 100 ng/ml ATc
We also measured cell death by analyzing the amount of mono- and oligonucleosomes in the cytosol (see Materials and Methods). Again the cells with induced pro-caspase-3 in combination with MG-132 treatment showed maximal cell death, which was reduced upon suppression of pro-caspase-3 expression (Figure 9, lanes 7 and 9). The level of cell death was insignificant in the control cell line under all conditions.

Effect of MG-132 on DNA cleavage in SKOV3 stable transfectants. Clone SKK-1 (control-transfected) and clone SK-1 (pNRTIS-33 pro-caspase-3) were seeded in 96 well plates (103 cells/well) and grown in the absence or presence (100 ng/ml) of ATc for 5 days. Thereafter cells were incubated in the absence or presence (10 μM) of MG-132, as indicated. Cell lysates were analysed in streptavidin-coated microtitre plates in the presence of antibodies directed against DNA and histones. The presence of mono- and oligonucleosomes was determined photometrically in the presence of substrate (see Materials and Methods)
Analysis of the effect of proteasome inhibitors on the OVCAR3 cell line overexpressing pro-caspase-3 showed similar results to those for the SKOV3 cells. Caspase-3-like activity was about 10-fold higher in clone TA with induced pro-caspase-3 expression in the presence of MG-132 (Figure 10, lanes 9 and 10). Similar results to those for SKOV3 were found for OVCAR3 by measurement of cell death (data not shown). The effect of taxol on caspase-3-like activity was again less marked compared to the effect of the proteasome inhibitors (data not shown), which indicates that taxol is a less potent activator of pro-caspase-3.

Effect of MG-132 on caspase-3-like activity in OVCAR3 stable transfectants. Clone TK (contro-transfected) and clone TA (pNRTIS-33pro-caspase-3) were grown in the absence or presence (100 ng/ml) of ATc for 5 days and were incubated with 5 or 10 μM MG-132 as indicated. Cells were lysed and caspase-3 like activity was measured against colorimetric substrate-Ac-DEVD-pNA. Lane 1, substrate alone; lanes 2–13 substrate plus cytoplasmic extracts from cells treated as indicated
Discussion
The apoptotic mechanisms involved in the regulation of cell fate in physiological or pathological conditions have been investigated extensively over many years. Several reports have shown that tumour cells have well developed ‘survival’ systems which include reduced responses to apoptotic stimuli.2,6,25
Recently, different approaches have been used to exploit apoptosis-inducing genes for gene therapy of cancer. Adenovirus-mediated inhibition of NF-κB function rendered melanoma cells susceptible to the cytotoxic effect of TNF.26 Overexpression of the Bax protein in A253 squamous cell carcinoma cells has been reported to result in an increased sensitivity to various chemotherapeutic agents.27 Adenoviral transfer of the pro-apoptotic gene Bak induced marked apoptosis in cancer cells in vitro and in vivo.28 Overexpression of the death-promoting Bcl-2 homologue Bik/Nbk can enhance drug-induced apoptosis in T-cell leukaemia H9 cells.29
Proteolytic cleavage of various cellular proteins, triggered mainly by the caspases, is a key element in apoptosis. The main players in this process are the effector caspases-3, -6 and -7. Recently, it has been shown that these caspases can also be effective tools for cancer gene therapy. Adenovirus-mediated transfer of pro-caspase-7 causes apoptosis in LNCaP cells.30 Coinfection with an adenovirus expressing pro-caspase-3 and an adenovirus expressing Fas ligand induced apoptosis in U-373MG glioma cells.17 In an AH130 liver tumour model, transduction of human pro-caspase-3 induced extensive apoptosis and reduced tumour volume when combined with etoposide administration.16
In this study we have demontrated the key role of pro-caspase-3 in regulation of apoptosis induced by proteasome inhibitors. In previous reports there has been controversy over whether overexpression of pro-caspase-3 alone can induce apoptosis.16,17,31 However, our transient transfection experiments in 293 cells and the data from the inducible ovarian cancer cell stable lines clearly showed that pro-caspase-3 overexpression is not sufficient to induce cell death. At the same time, the level of pro-caspase-3 appears to play a crucial role in the cellular response to treatment with different drugs.
It has been shown that most anti-cancer drugs can induce apoptosis, mainly by activation of the effector caspases-3, -6 and -7.32,33 However, data from MCF-7 cells and analysis of caspase-3 knockout mice have shown that caspase-3 deficiency cannot block the caspase cascade.34 Identification of specific activators of different caspases could be useful for caspase-based gene therapy of cancer.
Here we have described a model based on a tetracycline-inducible system for caspase expression to identify drugs which can specifically induce pro-caspase-3 activation and subsequent cellular apoptosis. Based on previous reports18,19,20,35,36,37,38 we tested proteasome inhibitors as potential potent activators of pro-caspase-3. In our ovarian cancer cell model we found that the apoptosis induced by the proteasome inhibitors MG-132, ALLN and lactacystin is regulated by the level of pro-caspase-3. We have shown that cell death can be enhanced when proteasome inhibitors are combined with pro-caspase-3 overexpression. Many drugs used for cancer treatment have been shown to induce apoptosis by activation of caspase-3,39,40,41,42 including taxol.32 Comparison between apoptosis induced by proteasome inhibitors and taxol in our ovarian cell line models revealed that proteasome inhibitors are more potent caspase-3 activators than taxol. These observations may be explained by a significant difference in the mechanism of cell death induced by these drugs. Taxol has been shown to induce apoptosis by stabilising microtubules, arresting the cells in G2/M phase with activation of JNK and reduction in basal p38 MAPK activity.23,43,44 In contrast, proteasome inhibitors can block protein turnover by inhibiting proteasome function and this leads to arrest of the cells in G2/M and induction of apoptosis by activation of caspase-3- and -6-like proteases.
Understanding mechanisms of apoptosis will help us develop strategies for the genetic treatment of cancer. Recent reports have demonstrated that drugs specifically blocking proteasome function are broadly active against multiple tumour cell types in vitro and in vivo.21,45 Our data strongly suggest that the combination of pro-caspase-3 overexpression with proteasome inhibitors can sensitise ovarian cancer cells to apoptosis. By using a suitable delivery system this approach could be applied for gene therapy of ovarian and other cancers.
Materials and Methods
Materials
Taxol (Paclitaxel) and proteasome inhibitors MG-132 (N-CBZ-leu-leu-leucinal) and Calpain inhibitor I, ALLN (N-acetyl-leu-leu-norleucinal) were purchased from Sigma. Lactacystin was purchased from Calbiochem (UK). Colorimetric substrate Ac-DEVD-pNA and fluorogenic substrate Ac-DEVD-AMC were purchased from Calbiochem (UK). Fluorogenic substrates Ac-YVAD-AMC and Ac-VEID-AFC were purchased from Pharmingen (Europe). Polyclonal goat anti-caspase-3 antibody was from Santa Cruz (Autogen Bioclear UK) and polyclonal rabbit anti-caspase-3 antibody (recognising the active form of caspase-3) was from Pharmingen (Europe). Affinity-purified mouse monoclonal antibody against poly (ADP-ribose) polymerase (PARP) was from Santa Cruz (Autogen Bioclear UK).
Cell lines
Human embryonal kidney (HEK) cells and the ovarian cancer cell line SKOV3 were grown in Dulbecco's modification of Eagle's minimal essential medium (DMEM) with 10% feotal calf serum (FCS). The ovarian cancer cell line OVCAR3 was grown in RPMI 1640 and supplemented with serum as described above.
Plasmids and transient transfection
Full-length human pro-caspase-3 cDNA was generated by RT–PCR from OVCAR3 cells and cloned into the mammalian expression vector pcDNA3 (Invitrogen) and the tetracycline-regulatable tricistronic vector pNRTIS-33 (neo-resistance gene and tetracycline-inhibited transactivator are within the tetracycline-responsive cassette).24 Constitutively active caspase-3 (reverse caspase-3) was kindly provided from Dr ES Alnemri.46 HEK 293 cells were transiently transfected with plasmids using a Calcium Phosphate Transfection kit (Invitrogen).
Generation of stable cell lines
SKOV3 and OVCAR3 were transfected with pNRTIS-33 pro-caspase-3 or pNRTIS-33 using SuperfectTM (Qiagen) as described by the manufacturer. The G-418 (1 mg/ml) selection in DMEM-10% FCS medium for SKOV3 and in RPMI1640-10% FCS for OVCAR3 cells was started 48 h after transfection. Several clones were isolated and tested for inducible expression of pro-caspase-3.
Flow cytometry
HEK 293 cells were grown in DMEM-10% FCS in the absence or presence of proteasome inhibitor MG-132 (10 μM) for 24 h and 48 h. Cell cycle status was analyzed with a FACSCalibur flow cytometer (Becton Dickinson) by measuring fluorescence from cells stained with propidium iodide.
Caspase-1-like assay
The caspase-1-like activity assay was performed with the fluorometric substrate Ac-YVAD-AMC in accordance with the protocol supplied by the manufacturer (Pharmingen). In brief, cells (106) were lysed in lysis buffer [10 mM Tris-HCl, 10 mM NaH2PO4/NaHPO4, pH 7.5, 130 mM NaCl, 1% Triton X-100, 10 mM NaPPi] on ice for 5–10 min, then centrifuged at 10 000×g for 15 min and then the supernatant was used for the protease assay. For each reaction 50 μl of cell lysate was used, with 20 μM (final concentration) Ac-YVAD-AMC and protease assay buffer [20 mM HEPES (pH 7.5), 10% glycerol, 2 mM DTT] to 1 ml. The reaction mixtures were incubated for 2 h at 37°C and the AMC liberated from Ac-YVAD-AMC was measured with a spectofluorometer at an excitation wavelength of 380 nm and an emission wavelength of 450 nm.
Caspase-3-like assay
Caspase-3-like activity was measured using the colorimetric substrate Ac-DEVD-pNA as has been described,20 or using the fluorometric substrate Ac-DEVD-AMC as described above for caspase-1-like assay.
Caspase-6-like assay
Caspase 6-like activity assay was performed using the fluorometric substrate Ac-VEID-AFC in accordance with the protocol supplied by the manufacturer (Pharmingen). In brief, cells were lysed in lysis buffer (see above for caspase-1-like assay) on ice for 5–10 min, then centrifuged at 10 000 g for 15 min and then the supernatant was used for the protease assay. For each reaction 50 μl of cell lysate was used, with 20 μM (final concentration) Ac-VEID-AFC and protease assay buffer [20 mM PIPES, 100 mM NaCl, 10 mM DTT, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, (pH 7.2)]. The reaction mixtures were incubated for 1 h at 37°C and the AFC liberated from Ac-VEID-AFC was measured by spectrofluorometer with an excitation wavelength of 400 nm and an emission wavelength of 505 nm.
Cell death assay
Apoptotic cell death was measured by using Cell Death Detection ELISAPLUS kit (Roche) exactly as recommended by the manufacturer. In brief SKOV3 and OVCAR3 stable cell lines were seeded in 96-well plates (103 cell/well) and grown in the presence or absence of 100 ng/ml ATc for 5 days. The cells were treated with the various drugs for 12 h and thereafter lysed in 200 μl lysis buffer for 30 min at room temperature. The lysates were centrifuged at 200×g for 10 min and 20 μl from the supernatant was transferred to streptavidin-coated microtitre plates (MTP) for analysis. To each well 80 μl of the immunoreagent containing monoclonal antibodies directed against DNA and histones was added and the plates were incubated on a shaker (300 r.p.m.) for 2 h at room temperature. The solution was then removed and the wells were rinsed three times with Incubation buffer. To each well was added 100 μl of substrate and after 10–20 min the amount of cytosolic mono- and oligonucleosomes (indication of cell death) was determined spectrophotometrically at 405 nm against substrate solution as a blank.
Immunoblot analysis
Cells were lysed in 1% Triton buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 10% Glycerol, 1% Triton X-100) plus the protease inhibitor cocktail CompleteTM (Boehringer Mannheim). After centrifugation at 13 000×g for 15 min at 4°C, supernatant was recovered. Cell lysates were subjected to SDS-polyacrylamide gel electrophoresis (SDS–PAGE) and the resolved proteins were transferred onto Immobilon-P membranes (Millipore) for probing with the various primary antibodies. Antigen-antibody complex was visualised using the ECLTM system (Amersham).
Abbreviations
- ALLN:
-
calpain inhibitor I
- ATc:
-
anhydrotetracycline
- PARP:
-
poly (ADP-ribose) polymerase
- Ac-DEVD-pNA:
-
acetyl-Asp-Glu-Val-Asp-p-nitroaniline
- Ac-YVAD-AMC:
-
acetyl-Tyr-Val-Ala-Asp-aminomethylcoumarin
- Ac-DEVD-AMC:
-
acetyl-Asp-Glu-Val-Asp-aminomethylcoumarin
- Ac-VEID-AFC:
-
acetyl-Val-Glu-Ile-Asp-aminotrifluoromethylcoumarin
- taxol (paclitaxel):
-
baccatin III N-benzyl-β-phenylisoserine ester
References
Kennedy SG, Kandel ES, Cross TK, Hay N . 1999 Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria Mol. Cell Biol. 19: 5800–5810
Krasilnikov MA . 2000 Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malignant transformation Biochemistry (Mosc). 65: 59–67
Leverrier Y, Thomas J, Mathieu AL, Low W, Blanquier B, Marvel J . 1999 Role of PI3-kinase in Bcl-X induction and apoptosis inhibition mediated by IL-3 or IGF-1 in Baf-3 cells Cell Death Differ. 6: 290–296
Tang C, Willingham MC, Reed JC, Miyashita T, Ray S, Ponnathpur V, Huang Y, Mahoney ME, Bullock G, Bhalla K . 1994 High levels of p26BCL-2 oncoprotein retard taxol-induced apoptosis in human pre-B leukemia cells Leukemia 8: 1960–1969
Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R . 1995 Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo Cancer Res 55: 4438–4445
Reed JC . 1997 Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies Semin. Hematol. 34: 9–19
Gazitt Y, Rothenberg ML, Hilsenbeck SG, Fey V, Thomas C, Montegomrey W . 1998 Bcl-2 overexpression is associated with resistance to paclitaxel, but not gemcitabine, in multiple myeloma cells Int. J. Oncol. 13: 839–848
Budihardjo I, Oliver H, Lutter M, Luo X, Wang X . 1999 Biochemical pathways of caspase activation during apoptosis Annu. Rev. Cell Dev. Biol. 15: 269–290
Juo P, Kuo CJ, Yuan J, Blenis J . 1998 Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade Curr. Biol. 8: 1001–1008
Rokhlin OW, Glover RA, Cohen MB . 1998 Fas-mediated apoptosis in human prostatic carcinoma cell lines occurs via activation of caspase-8 and caspase-7 Cancer Res. 58: 5870–5875
Shima Y, Nakao K, Nakashima T, Kawakami A, Nakata K, Hamasaki K, Kato Y, Eguchi K, Ishii N . 1999 Activation of caspase-8 in tranforming growth factor-beta-induced apoptosis of human hepatoma cells Hepatology 30: 1215–1222
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X . 1997 Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade Cell 91: 479–489
Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES . 1998 Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization Mol. Cell 1: 949–957
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ . 1999 Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner J. Cell Biol. 144: 281–292
Nicholson DW . 1999 Caspase structure, proteolytic substrates, and function during apoptotic cell death Cell Death Differ. 6: 1028–1042
Yamabe K, Shimizu S, Ito T, Yoshioka Y, Nomura M, Narita M, Saito I, Kanegae Y, Matsuda H . 1999 Cancer gene therapy using a pro-apoptotic gene, caspase-3 Gene Ther 6: 1952–1959
Shinoura N, Muramatsu Y, Yoshida Y, Asai A, Kirino T, Hamada H . 2000 Adenovirus-mediated transfer of caspase-3 with Fas ligand induces drastic apoptosis in U-373MG glioma cells Exp. Cell Res. 256: 423–433
Drexler HC . 1997 Activation of the cell death program by inhibition of proteasome function Proc. Natl. Acad. Sci. USA. 94: 855–860
Shinohara K, Tomioka M, Nakano H, Tone S, Ito H, Kawashima S . 1996 Apoptosis induction resulting from proteasome inhibition Biochem. J. 317: 385–388
Zhang XM, Lin H, Chen C, Chen BD . 1999 Inhibition of ubiquitin-proteasome pathway activates a caspase-3-like protease and induces Bcl-2 cleavage in human M-07e leukaemic cells Biochem. J. 340: 127–133
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ . 1999 Proteasome inhibitors: a novel class of potent and effective antitumor agents Cancer Res. 59: 2615–2622
Pucci B, Bellincampi L, Tafani M, Masciullo V, Melino G, Giordano A . 1999 Paclitaxel induces apoptosis in Saos-2 cells with CD95L upregulation and Bcl-2 phosphorylation Exp. Cell Res. 252: 134–143
Millenbaugh NJ, Gan Y, Au JL . 1998 Cytostatic and apoptotic effects of paclitaxel in human ovarian tumors Pharm. Res. 15: 122–127
Tenev T, Bohmer SA, Kaufmann R, Frese S, Bittorf T, Beckers T, Bohmer FD . 2000 Perinuclear localization of the protein-tyrosine phosphate SHP-1 and inhibition of epidermal growth factor-stimulated STAT1/3 activation in A431 cells Eur. J. Cell Biol. 79: 261–271
Reed JC . 1999 Dysregulation of apoptosis in cancer J. Clin. Oncol. 17: 2941–2953
Bakker TR, Reed D, Renno T, Jongeneel CV . 1999 Efficient adenoviral transfer of NF-kappaB inhibitor sensitizes melanoma to tumor necrosis factor-mediated apoptosis Int. J. Cancer 80: 320–323
Guo B, Cao S, Toth K, Azrak RG, Rustum YM . 2000 Overexpression of Bax enhances antitumor activity of chemotherapeutic agents in human head and neck squamous cell carcinoma Clin. Cancer Res. 6: 718–724
Pataer A, Fang B, Yu R, Kagawa S, Hunt KK, McDonnell TJ, Roth JA, Swisher SG . 2000 Adenoviral Bak overexpression mediates caspase-dependent tumor killing Cancer Res. 60: 788–792
Daniel PT, Pun KT, Ritschel S, Sturm I, Holler J, Dorken B, Brown R . 1999 Expression of the death gene Bik/Nbk promotes sensitivity to drug-induced apoptosis in corticosteroid-resistant T-cell lymphoma and prevents tumor growth in severe combined immunodeficient mice Blood 94: 1100–1107
Marcelli M, Cunningham GR, Walkup M, He Z, Sturgis L, Kagan C, Mannucci R, Nicoletti I, Teng B, Denner L . 1999 Signaling pathway activated during apoptosis of the prostate cancer cell line LNCaP: overexpression of caspase-7 as a new gene therapy strategy for prostate cancer Cancer Res. 59: 382–390
Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC . 1998 IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs Cancer Res. 58: 5315–5320
Weigel TL, Lotze MT, Kim PK, Amoscato AA, Luketich JD, Odoux C . 2000 Paclitaxel-induced apoptosis in non-small cell lung cancer cell lines is associated with increased caspase-3 activity J. Thorac. Cardiovasc. Surg. 119: 795–803
Kaufmann SH, Earnshaw WC . 2000 Induction of apoptosis by cancer chemotherapy Exp. Cell Res. 256: 42–49
Kurokawa H, Nishio K, Fukumoto H, Tomonari A, Suzuki T, Saijo N . 1999 Alteration of caspase-3 (CPP32/Yama/apopain) in wild-type MCF-7, breast cancer cells Oncol. Rep. 6: 33–37
Drexler HC, Risau W, Konerding MA . 2000 Inhibition of proteasome function induces programmed cell death in proliferating endothelial cells FASEB. J. 14: 65–77
Fujita E, Mukasa T, Tsukahara T, Arahata K, Omura S, Momoi T . 1996 Enhancement of CPP32-like activity in the TNF-treated U937 cells by the proteasome inhibitors Biochem. Biophys. Res. Commun. 224: 74–79
Imajoh-Ohmi S, Kawaguchi T, Sugiyama S, Tanaka K, Omura S, Kikuchi H . 1995 Lactacystin, a specific inhibitor of the proteasome, induces apoptosis in human monoblast U937 cells Biochem. Biophys. Res. Commun. 217: 1070–1077
Pasquini LA, Besio Moreno M, Adamo AM, Pasquini JM, Soto EF . 2000 Lactacystin, a specific inhibitor of the proteasome, induces apoptosis and activates caspase-3 in cultured cerebellar granule cells J. Neurosci. Res. 59: 601–611
Motwani M, Delohery TM, Schwartz GK . 1999 Sequential dependent enhancement of caspase activation and apoptosis by flavopiridol on paclitaxel-treated human gastric and breast cancer cells Clin. Cancer Res. 5: 1876–1883
Zhao EG, Song Q, Cross S, Misko I, Lees-Miller SP, Lavin MF . 1998 Resistance to etoposide-induced apoptosis in a Burkitt's lymphoma cell line Int. J. Cancer 77: 755–762
Turnbull KJ, Brown BL, Dobson PR . 1999 Caspase-3-like activity is necessary but not sufficient for daunorubicin-induced apoptosis in Jurkat human lymphoblastic leukemia cells Leukemia 13: 1056–1061
Henkels KM, Turchi JJ . 1999 Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines [published erratum appears in Cancer Res 2000 Feb 15; 60(4): 1150] Cancer Res. 59: 3077–3083
Stone AA, Chambers TC . 2000 Microtubule inhibitors elicit differential effects on MAP kinase (JNK, ERK and p38) signaling pathways in human KB-3 carcinoma cells Exp. Cell Res. 254: 110–119
Wang TH, Popp DM, Wang HS, Saitoh M, Mural JG, Henley DC, Ichijo H, Wimalesena J . 1999 Microtubule dysfunction induced by paclitaxel initiates apoptosis through both c-Jun N-terminal kinase (JNK)-dependent and -independent pathways in ovarian cancer cells J. Biol. Chem. 274: 8208–8216
You SA, Basu A, Haldar S . 1999 Potent antitumor agent proteasome inhibitors: a novel trigger for Bcl2 phoshporylation to induce apoptosis Int. J. Oncol. 15: 625–628
Srinivasula SM, Ahmad M, MacFarlane M, Luo Z, Huang Z, Fernandes-Alnemri T, Alnemri ES . 1998 Generation of constitutively active recombinant caspases-3 and -6 by rearrangement of their subunits J. Biol. Chem. 273: 10107–10111
Acknowledgements
We are grateful to Dr. Frank-D Bohmer for kindly providing the tri-cistronic tetracycline regulatable vector pNRTIS-33. We would like to thank Dr. ES Alnemri for providing reverse-caspase-3. We also thank Zoë Leech for technical assistance and Drs. Helen Hurst and Hani Gabra for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by SJ Martin
Rights and permissions
About this article
Cite this article
Tenev, T., Marani, M., McNeish, I. et al. Pro-caspase-3 overexpression sensitises ovarian cancer cells to proteasome inhibitors. Cell Death Differ 8, 256–264 (2001). https://doi.org/10.1038/sj.cdd.4400808
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400808
Keywords
This article is cited by
-
REG gamma: a potential marker in breast cancer and effect on cell cycle and proliferation of breast cancer cell
Medical Oncology (2011)
-
MG132 induced apoptosis is associated with p53-independent induction of pro-apoptotic Noxa and transcriptional activity of β-catenin
Apoptosis (2006)
-
Caspase activation and apoptosis in response to proteasome inhibitors
Cell Death & Differentiation (2005)
-
Procaspase 3 expression in ovarian carcinoma cells increases survivin transcription which can be countered with a dominant-negative mutant, survivin T34A; a combination gene therapy strategy
Oncogene (2003)
-
Induction of apoptosis in human osteosarcoma Saos-2 cells by the proteasome inhibitor MG132 and the protective effect of pRb
Cell Death & Differentiation (2003)
