
Abstract
The aim of the present study was to characterize expression and mutation of p53 during the neoplastic progression from Barrett's esophagus to adenocarcinoma and to test the reliability of immunohistochemistry for p53 overexpression as an indicator of p53 mutation in this context. The association of both gene mutation and protein accumulation with clinicopathological findings and survival was also studied. A total of 77 samples from 30 esophagectomy specimens with Barrett's esophagus and adenocarcinoma of patients in longitudinal clinical follow-up were analyzed. Different lesions (intestinal metaplasia, dysplasia, and adenocarcinoma) as well as normal squamous-cell esophageal epithelia were sampled from formalin-fixed, paraffin-embedded tissues by microdissection. Mutations in p53 Exons 5 to 9 were detected by polymerase chain reaction–single-strand conformation polymorphisms (PCR-SSCP) and confirmed by direct DNA sequencing. Nuclear accumulation of p53 protein was analyzed immunohistochemically from tissue sections adjacent to those used for microdissection. p53 gene mutations were found in 17 and p53 protein accumulation were found in 20 tumor samples. Of the 17 adenocarcinomas with a p53 mutation, 16 stained positive for p53 protein. p53 mutations were detected significantly more frequently in high-grade dysplastic than in low-grade dysplastic lesions (77% versus 29%, P < 0.01). In contrast, nuclear accumulation of p53 was detected in 85% of high-grade and 71% of low-grade dysplastic lesions. In eight cases with p53 mutation, the mutation identified in the tumors was also detected in premalignant lesions, mainly in high-grade dysplasia. In four cases of p53-mutated tumors, clones with different p53 mutations were detected in premalignant lesions. Neither p53 mutations nor p53 protein accumulations were found in metaplastic lesions. In summary, we found that p53 mutations occurred mainly during the transition from low-grade to high-grade dysplasia in the neoplastic progression of Barrett's esophagus but not in the nondysplastic Barrett's mucosa. Mutational analysis of p53 by PCR-SSCP and p53 accumulation by immunohistochemistry were mostly concordant in adenocarcinoma and high-grade dysplastic lesions but frequently discordant in low-grade dysplastic lesions. No correlation between p53 gene mutation or p53 accumulation and clinicopathological findings was observed in this study.
Similar content being viewed by others

INTRODUCTION
Barrett's esophagus represents a peculiar form of healing in the lining of the distal esophagus that occurs in response to chronic gastroesophageal reflux (1, 2). The most important clinical significance of this disease is the associated predisposition to adenocarcinoma of the esophagus and gastric cardia (3, 4). It is hypothesized that cancer does not arise in Barrett's esophagus de novo but as a result of progression from a metaplastic intestinal epithelium through increasing degrees of dysplasia and finally to invasive carcinoma (5, 6, 7, 8). An increased understanding of molecular alterations during this neoplastic progression might allow improvement of tumor control and prevention and eventually lead to better management of the disease.
The p53 tumor suppressor gene encodes a nuclear phosphoprotein, which functions as a transcription factor that controls the expression of many genes important in the regulation of the cell cycle. This phosphoprotein is also a factor in triggering apoptosis after certain types of genomic damage (9). Deletion of one allele in the short arm of Chromosome 17 and a functionally inactivating mutation of the other allele at the specific location are among the most common genetic abnormalities documented in human cancers (10). Numerous studies have found evidence to suggest that p53 inactivation contributes to the development of Barrett's esophageal adenocarcinoma. Casson et al. (11) first demonstrated p53 mutations in Barrett's adenocarcinoma and premalignant lesions. Later, p53 gene mutations were found to be common and to develop as relatively early events in the progression to cancer in Barrett's esophagus (12). These results have been reconfirmed by many other investigators (13, 14, 15). A close association between 17p allelic loss and p53 gene mutation has also been found (16). Additional evidence for the involvement of p53 mutations in the neoplastic progression toward Barrett's adenocarcinoma comes from studies of p53 protein overexpression by means of multiparameter flow cytometry and immunohistochemistry (17, 18, 19). Some studies have correlated p53 alterations with clinicopathologic findings and outcomes (20, 21, 22). However, despite the presence of increasing amounts of information, the precise role of p53 alterations in the natural history from benign to malignant lesions in Barrett's and the potential clinical significance of these observation remain unclear (23).
Wild-type p53 protein has a short half-life, and the level present in the nucleus of normal cells is below the sensitivity of immunohistochemical detection. Missense mutations in p53 gene result in a protein with a prolonged half-life that then accumulates in the nucleus (24). The increased levels of p53 are because of metabolic stabilization rather than overexpression (25). p53 protein overexpression detected by immunohistochemistry (IHC) has been proposed as an indirect method of detecting a p53 mutation. It is relatively cheap, quick, and easy compared with other techniques, such as DNA sequencing or polymerase chain reaction single-strand conformation polymorphisms (PCR-SSCP). In addition, the technique is available in most pathology laboratories.
Studies on colorectal and ovarian cancer have shown that mutation of the p53 gene and overexpression of p53 protein are significantly correlated (26, 27). In contrast, these features are not correlated in lung or head and neck cancers (28). How p53 gene mutations and p53 immunoreactivity correlate in preneoplastic lesions of Barrett's esophageal mucosa and their associated cancers has not been extensively studied.
The aims of the present study were to characterize expression and mutation of p53 during the progression of Barrett's esophagus to adenocarcinoma; to test the reliability of immunohistochemistry (IHC) as an indicator of p53 mutation; and to evaluate the association of both gene mutation and protein accumulation with pathological findings and survival.
MATERIALS AND METHODS
Tissue Samples
Thirty esophagectomy specimens for adenocarcinoma developed in a Barrett's esophagus between 1986–1996 were selected from the files of the Institute of Pathology at the University of Lausanne, Lausanne, Switzerland. Patients had received neither radiation therapy nor chemotherapy before surgery. The age of the patients (27 men and 3 women) ranged from 46 to 86 years and averaged 65 years. Follow-up results were available for 25 patients. Samples were formalin fixed and paraffin embedded. All available histological sections, which had been collected by mapping of the lesions as described for gastric carcinomas (29), were re-examined by two pathologists (MCO and CF). Five lesions were selected from each esophagectomy specimen, which resulted in 77 samples encompassing 30 invasive adenocarcinomas, 17 metaplasias, 17 low-grade (LG) dysplasias, and 13 high-grade (HG) dysplasias. For each patient, a sample of normal squamous epithelium was also studied.
Diagnosis and Grading
Hematoxylin and eosin–stained slides were screened to identify the different lesions. Barrett's esophagus was diagnosed when metaplastic epithelium (columnar epithelium with goblet cells) was present above the macroscopically identified gastroesophageal junction. Dysplasia was graded into two categories, low grade and high grade, based on the degree of cytonuclear and architectural atypia (30). Tumors were graded into well-, moderately and poorly differentiated adenocarcinoma and the depth of invasion was defined according to the UICC TNM classification of malignant tumors, fifth edition 1997.
Detection of p53 Mutation by Nonradioactive SSCPs
After deparaffinization in xylol and methanol, esophageal adenocarcinoma and premalignant lesions were carefully removed from the sections by microdissection, as described elsewhere (31). DNA was prepared by digestion with proteinase K, treated with Chelex, extracted with phenol/chloroform, and then precipitated with ethanol (31). The methods for nonradioactive PCR-SSCP have been previously described in detail (32). Primers for Exons 5 to 9 of the p53 gene have also been described (33).
Direct DNA Sequencing
After SSCP, the mutation-specific band observed on 30% nondenaturing MDE gel was cut out and reamplified using the same primers. The PCR-amplified DNAs were purified using a QIAquick PCR purification Kit (Qiagen, Germany), and the purified product was sequenced on an ABI PRISM 310 automatic sequencer using the dideoxy dye termination method.
Immunohistochemical Analysis
All the selected tissue samples were examined by immunohistochemistry using the Strept ABComplex/HRP Duet Kit (DAKO, Denmark) and visualized with 3,3′-diaminobenzidine. The monoclonal mouse anti-human p53 protein (Clone DO-7, DAKO, Denmark), which is known to recognize an epitope between amino acids 19 to 26 and to react with wild-type and mutant p53 protein, was used in this study (diluted 1:500, 26.8 ug/mL). Two staining patterns were distinguished: positive, when more than 10% of nuclei were positive, in clusters or scattered through the tissue; and negative, when no or only a few cells (<10%) showed a nuclear staining.
Statistics
The χ2 test was used for univariate analysis, and unweighted logistic regression for the multivariate analysis.
RESULTS
p53 Mutation and Protein Accumulation in Adenocarcinomas
p53 gene mutations, confirmed by direct DNA sequencing, were found in 17 of the 30 (57%) adenocarcinomas (Table 1). In these tumors, p53 gene mutations were distributed as follows: seven in Exon 5, one in Exon 6, six in Exon 7, and three in Exon 8. No mutation was found in Exon 9. There were 15 point mutations and 2 deletions (one of 10 bp and another of 19 bp). G:C to A:T transition was found rather frequently (7/17, 41%), and among them, five occurred at CpG sites. Five G:C to T:A transversions (one of them resulting in a stop codon) were also present.
Tissue sections adjacent next to those used for microdissection were analyzed in the same region by IHC. p53 protein accumulation was found in 20 of the 30 adenocarcinomas (67%). Nuclear staining was characteristic, and no cytoplasmic staining was noted (Fig. 1A).

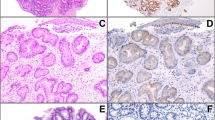
p53 protein overexpression and p53 mutation in a case with premalignant lesions and adenocarcinoma in Barrett's esophagus (Table 1, case 5). A, IHC using the DO-7 antibody (M): metaplasia showing absence immunostaining for p53; (LG), (HG) and (T): LG dysplasia, HG dysplasia, and moderately differentiated esophageal adenocarcinoma showing nuclear immunostaining for p53 (magnification, 100 ×). B, PCR-SSCP analysis of exon 7 of the p53 gene. Mobility observed in LG dysplasia differed from that in HG dysplasia and adenocarcinoma (T). No mutation was detected in metaplasia (M). DNA from normal tissues (N) of the same patient was used as a negative control.
Of 17 adenocarcinoma cases with p53 mutation, 16 also showed p53 protein accumulation. One case with a 10-bp deletion in the p53 gene, resulting in the production of a partially different p53 protein, showed no staining by IHC (Case 7). Nuclear accumulation of p53 protein was found in all 14 cases with missense mutation of the p53 gene. In 4 of the 13 cases which p53 mutations were negative by PCR-SSCP, p53 protein accumulation was observed.
p53 Mutation and Protein Accumulation in Premalignant Lesions
To investigate whether p53 mutations occurred consistently throughout the Barrett's epithelium, SSCP analysis was used to screen premalignant lesions (metaplasia, LG and HG dysplasia) sampled from all 17 cases in which a p53 mutation was found in the adenocarcinoma (Table 1). p53 mutations were detected significantly more frequently (P < 0.01) in HG-dysplastic (10/13; 77%) than in LG-dysplastic lesions (5/17; 29%). In 47% (8/17 cases) of tumors with a p53 mutation, the same mutation was also detected in at least one premalignant lesion. In four cases, the p53 mutation detected in the premalignant lesions differed from those in the associated adenocarcinomas. No p53 mutation was observed in metaplastic lesions.
p53 overexpression was detected in 85% (11/13) of specimens with HG dysplasia and in 71% (12/17) of those with LG dysplasia. No p53 protein accumulation was found in the 17 intestinal metaplastic lesions without dysplasia (Fig. 1).
Positive p53 staining was found in 13 of 15 (87%) premalignant lesions with a p53 mutation. The nonconcordant cases were two HG dysplastic lesions, one of which showed a 10-bp deletion that was also IHC negative in the tumor, and the other associated with an IHC-positive adenocarcinoma that had the same p53 mutation. Of the 15 dysplastic lesions negative by PCR-SSCP, 9 (60%) were p53 positive by IHC (Table 1). Most of them (7/9, 78%) were LG dysplasia (Fig. 1).
p53 Alterations and Clinicopathological Parameters
Table 2 provides the correlation between alteration of p53 and clinicopathological parameters. p53 mutation and p53 protein accumulation were analyzed according to patient gender, tumor histological type, pathologic stage, and survival. The incidence of p53 mutation and nuclear overexpression was higher in patients with lymph nodes metastasis, but the difference was not statistically significant. No significant differences were found according to the gender of patients or pathological characteristics of the specimens. The five patients lacking follow-up data were excluded from survival analysis. No differences in survival were observed between the patients with p53 mutations or p53 protein accumulation in esophageal adenocarcinoma and those without.
DISCUSSION
Using molecular and immunohistochemical techniques, we characterized the spectrum of p53 alterations during neoplastic progression in Barrett's esophagus and assessed their correlation with survival for patients with esophageal adenocarcinoma. In 30 cases of Barrett's esophageal adenocarcinoma, p53 mutations were found in 17 cases (57%), whereas p53 protein accumulation was detected in 20 cases (67%). These results correspond to the incidence of p53 alterations in Barrett's esophageal adenocarcinomas reported by Kubba et al. (23), who reviewed publications on the subject over the last 10 years. Previous studies have shown that the spectrum of p53 gene mutations is dependent on the cancer type (34, 35). Most of the mutations occur in Exons 5 to 9, which encode the DNA-binding domain (36). The variation found in p53 mutations concerns the position of mutational hot spots as well as the mutation type such as missense mutations (transition or transversion) and null mutations (nonsense, deletions, insertions, and splicing junction mutations). In esophageal squamous cell carcinoma as in lung carcinoma, a high frequency of G:C to T:A transversions has been observed (37, 38, 39). This pattern has been linked to cigarette smoking or the occupational exposure to polycyclic aromatic hydrocarbons (40). In Barrett's esophageal carcinoma, however, the mutation profile is different. In our series, in addition to G:C to T:A transversions (5/30 cases), G:C to A:T transitions were found frequently (7/30), and most of them occurred at CpG dinucleotides. Mutational hotspots at CpG dinucleotides may reflect endogenous mutagenic mechanisms, such as deamination of 5-methylcytosine to thymidine followed by a faulty repair procedure.
On histological section, the simplest and most practical approach is the use of IHC to detect p53 protein accumulation. Few studies have compared the results of p53 mutation analysis to p53 overexpression by IHC in esophageal adenocarcinoma. The reported discordance between these two approaches has ranged between 21 and 41% of cases (18, 41). The number of discordant cases in the present study (20%) is comparable to the data reported in the literature. We found that of 20 adenocarcinomas with p53 protein accumulation, 16 (80%) had p53 gene mutations. The discrepancy between IHC and mutation analysis was not due to sampling error: in this study, all of the IHC analysis was done in tissue section adjacent to those used for microdissection for p53 gene mutation analysis.
The results confirm that p53 gene mutation and/or p53 protein accumulation are common in Barrett's adenocarcinoma. One aim of this study was to determine the step at which p53 mutation occurs during neoplastic progression. p53 mutations were detected more frequently (P < 0.01) in HG dysplasia surrounding the tumor than in LG dysplasia. Furthermore, the mutations found in HG dysplasia were generally identical to those of the corresponding tumors, whereas different mutations were commonly detected in LG dysplasia. The finding of identical p53 mutations in HG dysplastic lesions and the associated tumors suggests that this genetic alteration occurs during the progression from LG to HG dysplasia. Some of the p53 mutations detected in the premalignant lesions, mainly in LG dysplasia, were different from those in adjacent adenocarcinomas. This indicates that, while the entire esophageal epithelium of an individual is exposed to dietary or environmental carcinogens, a lesion may arise from independent mutated cell clones at several different sites, some of which may eventually progress to carcinoma. It appears that p53 mutation is important in the later steps of the progression to carcinoma.
p53 overexpression occurred at a similar high frequency in the analyzed HG and LG dysplastic lesions. A high discordance rate between detection of p53 alteration by SSCP and IHC was observed in LG dysplastic lesions (7/17, 41%). The high incidence of p53 overexpression in LG dysplasia could be a consequence of the accumulation of wild-type p53 and/or the presence of p53 mutated clones, too small to be detected by SSCP analysis. In fact, only a few glands were positive by IHC in some LG dysplastic lesions in which no p53 mutations were detected. Neither p53 mutation nor p53 overexpression were observed in metaplastic lesions without dysplasia. It appears that in Barrett's esophagus p53 is not affected in the nondysplastic mucosa. These results indicate that p53 staining may have potential value for confirming a suspected diagnosis of LG dysplasia, as reported by other investigators (42).
The prognostic significance of p53 alterations in esophageal adenocarcinoma have been studied by many groups. Casson et al. (20) reported a series of 61 patients in which p53 alterations (gene mutation and protein accumulation) occurred mainly in poorly differentiated adenocarcinoma which showed significantly reduced postoperative survival. However, Duhaylongsod et al. (43) and Coggi et al. (44) failed to find any association between p53 gene mutation or protein accumulation and survival in patients with esophageal adenocarcinoma. In this study, we found no correlation between p53 mutation or nuclear overexpression and clinicopathological parameters. p53 alterations were not associated with a poorer patient survival rate. However, it should be emphasized that these findings are based on a relatively limited number of cases, and interpretation of results concerning the association between p53 alterations and prognosis need to be confirmed in larger prospective multicenter studies (20).
In conclusion, we found that p53 alterations are common and occur mainly during the transition from LG to HG dysplasia in the neoplastic progression of Barrett's esophagus. A minor discordance between p53 mutation and p53 accumulation was observed in tumor and HG dysplastic lesions, but this discordance was major in LG dysplastic lesions. Neither p53 mutation nor p53 protein accumulation had independent prognostic value in patients with Barrett's adenocarcinoma.
References
Phillips RW, Wong RKH . Barrett's esophagus. Natural history, incidence, etiology and complications. Gastroenterol Clin North Am 1991; 20: 791–816.
Spechler SJ, Zeroogian JM, Antonioli DA, Wang HH, Goyal RK . Prevalence of metaplasia at the gastro-oesophageal junction. Lancet 1994; 344: 1533–1536.
Van der Veen AH, Dees J, Blankenstein JD, Van Blankenstein M . Adenocarcinoma in Barrett's oesophagus: an overrated risk. Gut 1989; 30: 14–18.
Spechler SJ, Robbins AH, Rubins HB, Vincent ME, Heeren T, Doos WG, et al. Adenocarcinoma and Barrett's esophagus: an overrated risk? Gastroenterology 1984; 84: 927–933.
Spechler SJ . The columnar-lined esophagus. History, terminology and clinical issues. Gastroenterol Clin North Am 1977; 26: 455–466.
Hameeteman W, Tytgat GNJ, Houthoff HJ, van den Tweel JG . Barrett's esophagus: development of dysplasia and adenocarcinoma. Gastroenterology 1989; 96: 1249–1256.
Reid BJ, Blount PL, Rubin CE, Levine DS, Haggitt RC, Rabinovitch PS . Flow-cytometric and histological progression to malignancy in Barrett's esophagus: prospective endoscopic surveillance of a cohort. Gastroenterology 1992; 102: 1212–1219.
Rabinovitch PS, Reid BJ, Haggitt RC, Norwood TH, Rubin CE . Progression to cancer in Barrett's esophagus is associated with genomic instability. Lab Invest 1988; 60: 65–71.
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW . Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991; 51: 6304–6311.
Levine AJ . The p53 tumor-suppressor gene. N Engl J Med 1992; 326: 1350–1352.
Casson AG, Mukhopadhyay T, Cleary KR, Ro JY, Levin B, Roth JA . p53 gene mutation in Barrett's epithelium and esophageal cancer. Cancer Res 1991; 51: 4495–4499.
Bennett WP, Hollstein MC, Metcalf RA, Welsh JA, He A, Zhu SM, et al. p53 mutation and protein accumulation during multistage human esophageal carcinogenesis. Cancer Res 1992; 52: 6092–6097.
Neshat K, Sanchez CA, Galipeau PC, Blount PL, Levine DS, Joslyn G, et al. p53 mutation in Barrett's adenocarcinoma and high-grade dysplasia. Gastroenterology 1994; 106: 1589–1595.
Schneider PM, Casson AG, Levin B, Garewal HS, Hoelscher AH, Becker K, et al. Mutation of p53 in Barrett's esophagus and Barrett's cancer: a prospective study of ninety-eight cases. J Thorac Cardiovasc Surg 1996; 111: 323–333.
Wang LD, Hong JY, Qiu SL, Gao H, Yang CS . Accumulation of p53 protein in human esophageal precancerous lesions: a possible early marker for carcinogenesis. Cancer Res 1993; 53: 1783–1787.
Gleeson CM, Sloan JM, McGuigan JA, Ritchie AJ, Russell SE . Base transitions at CpG dinucleotides in the p53 gene are common in esophageal adenocarcinoma. Cancer Res 1995; 55: 3406–3411.
Ramel S, Reid BJ, Sanchez CA, Blount PL, Levine DS, Neshat K, et al. Evaluation of p53 protein expression in Barrett's esophagus by two-parameter flow cytometry. Gastroenterology 1992; 102: 1220–1228.
Hamelin R, Flejou JF, Muzeau F, Potet F, Laurent-Puig P, Fekete F, et al. TP53 gene mutation and p53 protein immunoreactivity in malignant and premalignant Barrett's esophagus. Gastroenterology 1994; 107: 1012–1018.
Krishnadath KK, Tilanus HW, Blankenstein MV, Bosman FT, Mulder AH . Accumulation of p53 protein in normal, dysplastic, and neoplastic Barrett's oesophagus. J Pathol 1995; 175: 175–180.
Casson AG, Tammemagi M, Eskandarian S, Redston M, McLaughlin J, Ozcelik H . p53 alteration in oesophageal cancer: association with clinicopathological features, risk factors, and survival. Mol Pathol 1998; 51: 71–79.
Casson AG, Manolopoulos B, Troster M, Kerkvliet N, O'Malley F, Inculet R, et al. Clinical implications of p53 gene mutation in the progression of Barrett's epithelium to invasive esophageal cancer. Am J Surg 1994; 167: 52–57.
Ireland AP, Clark GWB, Demeester TR . Barrett's esophagus: the significance of p53 in clinical practice. Ann Surg 1997; 225: 17–30.
Kubba AK, Pool NA, Watson A . Role of p53 assessment in management of Barrett's esophagus. Dig Dis Sci 1999; 44: 659–667.
Hall PA, Lane DP . p53 in tumour pathology: can we trust immunohistochemistry? J Pathol 1994; 172: 1–4.
Finlay CA, Hinds PW, Tan TH, Eliyahu D, Oren M, Levine AJ . Activating mutations for transformation by p53 produce a gene product that forms a hsc700-p53 complex with an altered half-life. Mol Cell Biol 1988; 8: 531–539.
Robbins BDP, Carrello S, House A, Iacopetta B . Comparison of p53 gene mutation and protein overexpression in colorectal carcinomas. Br J Cancer 1994; 70: 585–590.
Kihana T, Tsuda H, Teshima S, Okada S, Matsuura S, Hirohashi S . High incidence of p53 mutation in human ovarian cancer and its association with nuclear accumulation of p53 protein and tumor DNA aneuploidy. Jpn J Cancer Res 1992; 83: 978–984.
Melhem MF, Law JC, El-Ashmawy L, Johnson JT, Landreneau RJ, Srivastava S, et al. Assessment of sensitivity and specificity of immunohistochemical staining of p53 in lung and head and neck cancers. Am J Pathol 1995; 146: 1170–1177.
Mori M, Adachi Y, Kakeji Y, Korenaga D, Sugimachi K, Motooka M, et al. Superficial flat-type early carcinoma of the stomach. Cancer 1992; 69: 306–313.
Haggitt RC . Histopathology of reflux-induced esophageal and supraesophageal injuries. Am J Med 2000; 108: 109S–111S.
Baisse B, Bian YS, Benhattar J . Microdissection by exclusion and DNA extraction for multiple PCR analyses from archival tissue sections. Biotechniques 2000; 28: 856–862.
Chaubert P, Bautista D, Benhattar J . An improved method for rapid screening of DNA mutations by non-radioactive single-strand conformation polymorphism procedure. Biotechniques 1993; 15: 586.
Hurlimann J, Chaubert P, Benhattar J . p53 gene alterations and p53 protein accumulation in infiltrating ductal breast carcinomas: correlation between immunohistochemical and molecular biology techniques. Mod Pathol 1994; 7: 423–428.
Hollstein M, Sidransky D, Vogelstein B, Harris CC . p53 mutations in human cancers. Science 1991; 253: 49–51.
Harris CC, Hollstein M . Clinical implications of the p53 tumor suppressor gene. N Engl J Med 1993; 329: 1318–1327.
Cho Y, Gorina S, Jeffrey PD, Pavletich NP . Crystal structure of a p53 tumor suppressor DNA-complex: understanding tumorigenic mutations. Science 1994; 265: 346–355.
Wagata T, Shibagaki I, Imamura M, Shimada Y, Toguchida J, Yandell DW, et al. Loss of 17p, mutation of p53 gene, and overexpression of p53 protein in esophageal squamous cell carcinoma. Cancer Res 1993; 53: 846–850.
Shi ST, Yang GY, Wang LD, Xue ZH, Feng B, Ding W, et al. Role of p53 gene mutations in human esophageal carcinogenesis: results from immunohistochemical and mutation analyses of carcinomas and nearby non-cancerous lesions. Carcinogenesis 1999; 20: 591–597.
Lohmann D, Putz B, Reich U, Bohm J, Prauer H, Hofler H . Mutational spectrum of the p53 gene in human small-cell lung cancer and relation to clinicopathological data. Am J Pathol 1993; 142: 907–915.
Greenblatt MS, Bennett WP, Hollstein M, Harris CC . Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54: 4855–4878.
Zambetti GP, Levine AJ . A comparison of biological activities of wild-type and mutant p53. FASEB J 1993; 7: 855–865.
Khan S, Do KA, Kuhnert P, Pillay SP, Papadimos D, Conrad R, et al. Diagnostic value of p53 immunohistochemistry in Barrett's esophagus: an endoscopic study. Pathology 1998; 30: 136–140.
Duhaylongsod FG, Gottfried MR, Iglehart JD, Vaughn AL, Wolfe WG . The significance of c-erb B-2 and p53 immunoreactivity in patients with adenocarcinoma of the esophagus. Ann Surg 1995; 221: 677–684.
Coggi G, Bosari S, Roncalli M, Graziani D, Bossi P, Viale G, et al. p53 protein accumulation and p53 gene mutation in esophageal carcinoma. Cancer 1997; 79: 425–432.
Acknowledgements
This work was supported by grants from the Foundation 2000 (Lausanne, Switzerland) and by the Fondation pour la Lutte contre le Cancer (Zurich, Switzerland).
The authors thank Jean-Christophe Stehle for advice on immunohistochemical techniques and Simone Burki for her helpful photographic aid.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bian, YS., Osterheld, MC., Bosman, F. et al. p53 Gene Mutation and Protein Accumulation during Neoplastic Progression in Barrett's Esophagus. Mod Pathol 14, 397–403 (2001). https://doi.org/10.1038/modpathol.3880324
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880324
Keywords
This article is cited by
-
Targeted genetic and epigenetic profiling of esophageal adenocarcinomas and non-dysplastic Barrett’s esophagus
Clinical Epigenetics (2022)
-
Utility of ancillary studies in the diagnosis and risk assessment of Barrett's esophagus and dysplasia
Modern Pathology (2022)
-
Expression of p53, p63, podoplanin and Ki-67 in recurring versus non-recurring oral leukoplakia
Scientific Reports (2021)
-
Systematic review and meta-analysis of immunohistochemical prognostic biomarkers in resected oesophageal adenocarcinoma
British Journal of Cancer (2015)
-
Neoangiogenesis. Assessment in Esophageal Adenocarcinomas
Indian Journal of Surgery (2015)
