
Abstract
Pulmonary large-cell neuroendocrine carcinoma (LCNEC) is a newly proposed clinicopathologic entity; a few cases of LCNEC have been reported in other sites, such as the uterine cervix and the thymus. In the salivary glands, LCNEC is extremely rare and is not recognized as a specific entity in the World Health Organization classification. We retrospectively reviewed from our files 1675 cases of surgically resected primary parotid gland tumors and found 2 cases of LCNEC that fulfilled the criteria of pulmonary LCNEC. These cases occurred in 72- and 73-year-old men who had short histories of enlarging parotid gland tumors. The tumors were composed of large cells that exhibited organoid, solid, trabecular, and rosette-like growth patterns with a high mitotic rate and a conspicuous tendency for necrosis. The tumor cells were polygonal and characterized by a moderate nuclear:cytoplasmic ratio, coarse chromatin, and conspicuous nucleoli. Immunohistochemical examination revealed that the tumor cells were positive for six general neuroendocrine markers, cytokeratin, p53, bcl-2, epidermal growth factor receptor, and cyclin D1. Markedly reduced expressions of p21Waf1 and p27Kip1 were also noticed. The Ki-67 labeling index was more than 50% in both cases. One case showed loss of heterozygosity at TP53 accompanied by a p53 gene point mutation. Loss of heterozygosity at chromosome 9p21 was detected in both cases; one was accompanied by a p16 gene silent point mutation. Both patients died of the disease, with recurrence 5 months and 4 years after surgery, respectively. These findings indicate that LCNEC is a rare but distinct salivary gland tumor with highly aggressive biologic behavior. Multiple alterations of cell cycle regulators and tumor suppressor genes may play an important role in presenting the biologic characteristics of this rare parotid gland tumor.
Similar content being viewed by others

INTRODUCTION
Travis et al. (1) proposed a new, distinctive clinicopathologic entity of pulmonary neuroendocrine tumors, large-cell neuroendocrine carcinoma (LCNEC), defined as a poorly differentiated and high-grade neuroendocrine tumor that has several morphologic and biologic features between atypical carcinoid and small cell carcinoma. Although LCNEC is a rare neoplasm most commonly arising in the lung (1, 2, 3), a few cases of LCNEC have been reported in other sites, such as the uterine cervix (4), thymus (5), stomach (6), and urinary bladder (7). In the salivary glands, LCNEC is extremely rare, and only two cases have been reported (8, 9). Therefore, the biologic behavior and molecular alterations of salivary gland LCNEC are still poorly understood.
We retrospectively reviewed from our files 1675 cases of primary parotid gland tumors that were surgically resected between 1953 and 1999 and found 2 cases of LCNEC that fulfilled the criteria of pulmonary LCNEC defined by Travis et al. (1). We present the clinicopathologic features and immunohistochemical and molecular-genetic characteristics of these two tumors, including gene alterations of p53, p16, and K-ras. To our knowledge, this is the first molecular-genetic assessment of LCNEC arising in the salivary gland.
MATERIALS AND METHODS
Case Selection and Tissue Preparation
From our review of 1675 parotid gland tumors, we selected 2 cases of LCNEC from among 13 cases that initially had been diagnosed as large cell–type undifferentiated carcinoma (10). Because the other 11 cases exhibited no neuroendocrine characteristics either morphologically or immunohistochemically (negative for both chromogranin A and synaptophysin), we excluded them from this study. Tissue sections fixed in 10% buffered formalin were processed by routine histologic techniques, including staining with periodic acid-Schiff, alcian blue, mucicarmine, Fontana-Masson, and Grimelius. In one case, the material was fixed for electron microscopy in 2.5% glutaraldehyde in phosphate buffer at 4° C, postfixed in 1% osmium tetroxide, and embedded in Epon-812. Ultrathin sections were stained with lead citrate and examined in a JEM-1200EX electron microscope (JEOL, Tokyo, Japan).
Immunohistochemistry
The deparaffinized and rehydrated slides were boiled in 10 mm citrate buffer (pH 6.0) for 20 min in a microwave oven. After cooling for 30 min, they were incubated with the primary antibody for 15 h at 4° C. The primary antibodies used in this study are listed in Table 1. The labeled streptavidin-biotin peroxidase method (LSAB Kit; DAKO, Glostrup, Denmark) was used for detection; 3,3′-diaminobenzidine was the chromogen. The sections were slightly counterstained with hematoxylin. The percentage of Ki-67–positive cells was determined by counting at least 1000 tumor cells, and then was recorded as the Ki-67 labeling index.
Molecular Analysis
DNA was extracted from the paraffin-embedded sections by a microdissection method. The primer sequences for polymerase chain reaction (PCR) amplification are listed in Table 2. Forward primers of TP53, IFNA, and D9S171 were labeled with 6-FAM for loss of heterozygosity (LOH) analysis. LOH analysis was performed with a fluorescence-based microsatellite PCR technique using an ABI PRISM 310 Genetic Analyzer (Perkin Elmer, Foster City, CA) and Gene Scan 2.01 software (Perkin Elmer). The ratios of the normal and tumor allele peak areas were compared for LOH assessment. A change in the ratio by more than 50% indicated the loss of one allele in the carcinoma DNA (11). Mutational analysis was performed for p53 exon 5–8, p16 exon 1 and 2, and K-ras exon 1 and 2 by direct DNA sequencing. The DNA was sequenced with a dRhodamine Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer) and an ABI PRISM 310 Genetic Analyzer.
RESULTS
Clinical Histories
Case 1
A 72-year-old man had a 4-month history of an enlarging right-sided parotid mass. Physical examination demonstrated a fist-sized mass in the right parotid area. A computed tomographic scan showed a solid parotid gland mass, 7 cm in diameter, with a necrotic, low-density area in the central portion. No metastatic foci were detected. Fine-needle aspiration cytology showed loosely cohesive tumor cells in the necrotic background (Fig. 1); these had large and pleomorphic nuclei with prominent nucleoli and coarse chromatin. The nuclear:cytoplasmic ratio of the tumor cells was moderate. The cytologic diagnosis was large cell undifferentiated carcinoma. After chemotherapy and 40 Gy radiation, total right parotidectomy with radical neck dissection was performed. Postoperatively, the patient received 20 Gy radiation. Four months later, local recurrence was found by fine-needle aspiration cytology in the right parotid gland area. Despite extensive chemoradiotherapy, the patient died of the disease 5 months after surgery.

Case 1. Fine-needle aspiration cytology, showing loosely cohesive tumor cells. The tumor cells have large and pleomorphic nuclei with prominent nucleoli and coarse chromatin. The nuclear:cytoplasmic ratio of the tumor cells is moderate (Papanicolaou stain).
Case 2
A 73-year-old man had discovered a mass the size of the tip of a small finger in the right parotid gland 4 months before admission. On examination, there was a firm mass measuring 3.3 × 3.0 × 2.7 mm in the right parotid area. No other primary sites were identified. A superficial right parotidectomy with radical neck dissection was done, and the patient received 40 Gy postoperative radiotherapy. Two years later, the tumor recurred in the right cervical lymph nodes. Although the patient received chemotherapy and 60 Gy additional radiation, he died of the disease 4 years after surgery.
Histopathologic Findings
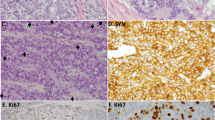
The tumors were composed of large cells that exhibited solid (Fig. 2A), organoid (Fig. 3A), and trabecular growth patterns. In Case 2, the tumor showed a lobular and nodular arrangement; in both cases, focal peripheral palisading was observed (Fig. 3B). Rosette-like patterns were easily found in Case 2 (Fig. 3C) but were rare in Case 1. Round tumor nests with central necrosis were common in both cases (Fig. 2A). No ductal or squamous differentiation was observed. A markedly desmoplastic stroma was evident in Case 1. Lymphoid infiltration was scant. The mitotic counts of the tumor cells were 70 and 82 per 10 high-power fields in Cases 1 and 2, respectively. Apoptotic cells were numerous. The tumor cells were large, with a moderate amount of mildly eosinophilic cytoplasm, and their nuclei displayed coarse and vesicular chromatin distribution with one or two prominent nucleoli (Figs. 2B and 3C). These cells were polygonal, and some were spindle shaped. No mucinous cells were detected by mucin stainings. In Case 1, scattered carcinoma cells displayed argyrophil granules on Grimelius staining, whereas Fontana-Masson staining was negative in both cases. No other coexisting salivary gland tumors were identified. In both cases, the tumor cells had invaded the adipose and muscular tissues beyond the parotid gland, and there was overt perineural and vascular involvement. Lymph node metastases in the neck region were also seen.

Case 1. A, large-cell neuroendocrine carcinoma displaying a solid growth pattern with massive necrosis (upper right). B, high-power view. The tumor cells have large and polygonal nuclei with vesicular chromatin and one or two prominent nucleoli. Two mitoses are seen in this figure (arrows) (hematoxylin and eosin stain).

Case 2. A, large-cell neuroendocrine carcinoma showing organoid growth pattern. B, solid growth with peripheral palisading of the tumor cells is seen. C, large tumor cells displaying a rosette-like structure (hematoxylin and eosin stain).
Immunohistochemical Findings
The results of the immunohistochemical stainings are summarized in Table 1. In both cases, the tumors were positive for six conventional neuroendocrine markers (1) to varying degrees. Chromogranin A was focal but intensely positive only in Case 2 (Fig. 4A), whereas in Case 1 diffuse and granular cytoplasmic immunoreactivity was seen for synaptophysin (Fig. 4B). Neuron-specific enolase and protein gene product 9.5 (Fig. 4C) were diffusely positive, but only focal immunoreactivity was found for CD57 (Leu-7), neural cell-adhesion molecule, and S-100 protein in both cases. The tumors also showed diffuse and strong immunostaining for cytokeratin (CK; AE1/AE3, CAM5.2) and epithelial membrane antigen. No immunoreactivity for CK20 was found in either case. Both cases demonstrated diffuse immunostaining for epithelial growth factor receptor and bcl-2. Abnormal and strong p53 nuclear staining (Fig. 4D) and nuclear and cytoplasmic p16 staining were diffusely distributed in both cases. Intense cyclin D1 nuclear immunoreactivity was observed in both cases. In Case 1, p21Waf1 and p27Kip1 were negative, whereas these were focally positive in Case 2. In both cases, a high cell-proliferative activity was demonstrated by the Ki-67 labeling index: 57.1% in Case 1 and 53.4% in Case 2.

A, Case 2. Focal but intensely chromogranin A–positive cells are seen. B, Case 1. Dot-like intracytoplasmic synaptophysin-immunoreactive cells are diffusely distributed. C, Case 2. The tumor cells are diffusely positive for protein gene product 9.5. D, Case 1. Strong positive staining for p53 can be observed in the nuclei of the tumor cells.
Ultrastructural Findings
Only Case 1 was available for ultrastructural examination. A few membrane-bound, dense-core neuroendocrine granules, ranging from 50 to 200 nm, were found in the scattered tumor cells (Fig. 5). The tumor cells were connected to each other by prominent desmosome-like junctions. Tonofilaments were inconspicuous, and no glandular lumens were identified.

Case 1. Electron micrograph of a tumor cell, showing the desmosome-like junctions (arrowheads) and a small group of membrane-bound, dense granules of the neurosecretory type (arrow) (scale: bar = 1 μm). Inset, Detail of neurosecretory granules (scale: bar = 200 nm).
Molecular Analysis
LOH assessment was performed on microsatellite loci of TP53 and chromosome 9p21 (IFNA and D9S171, flanking the p16 locus). In both cases, D9S171 was not informative (homozygous) but the other two loci were informative (heterozygous). LOH was detected in Case 1 at TP53 (Fig. 6A) and in both cases at IFNA (Fig. 6B). For the analysis of p53 mutations, we amplified and sequenced four highly conserved gene regions, corresponding to exons 5 through 8. Only Case 1 showed point mutation: CAT to AAT transition in codon 179 on exon 5, resulting in a His substitution for Asn (Fig. 7). Case 2 demonstrated a silent mutation: GGG (Gly) to GGT (Gly) at codon 127 in exon 2 of p16. No p16 mutations were found in Case 1. No point mutations were detected at the K-ras codon 12, 13, and 61 in both cases.

Fluorescent loss of heterozygosity analysis. A, Case 1 (TP53). Shorter allele (119 bp) showing 87% signal reduction in carcinoma tissue. B, Case 2 (IFNA). Longer allele (148 bp) showing 77% signal reduction in carcinoma tissue. Both the TP53 and the IFNA genes indicate loss of heterozygosity in the carcinoma tissue of Case 1 and Case 2, respectively.

Case 1. Direct sequencing analysis of polymerase chain reaction product of p53 exon 5. CAT to AAT transition in codon 179 results in a His substitution for Asn (arrow).
DISCUSSION
Undifferentiated carcinomas of the salivary glands are uncommon tumors with high-grade malignancy. They are generally subclassified into three groups: small (usually called simply small cell carcinoma) and large cell types of undifferentiated carcinomas (8, 10) and undifferentiated carcinomas with lymphoid stroma (12). Small cell carcinomas of the salivary glands, many of which show neuroendocrine differentiation (13, 14, 15, 16), are well-defined tumors that are included in the World Health Organization classification of salivary gland tumors (17), although the incidence is low (approximately 1 to 2% of all major salivary gland malignancies [18]). Salivary gland neuroendocrine carcinomas other than the small cell type have received little attention and are not recognized as a specific entity in the World Health Organization classification (17). Hui et al. (8) found that one of their four cases of large cell–type undifferentiated carcinoma of the salivary glands ultrastructurally had neuroendocrine granules, but they did not regard this case as a separate entity. Our LCNEC cases initially had been miscategorized as large cell–type undifferentiated carcinomas but showed marked necrosis and a high mitotic rate. These tumors seem to be distinct from large cell–type undifferentiated carcinomas, based in particular on the presence of the neuroendocrine pattern, including the rosette-like structures and the organoid and peripheral palisading growth patterns, as well as the immunohistochemical or electron microscopic features. Therefore, our cases constitute a rare type of salivary gland tumor, LCNEC, which can be regarded as a distinct entity arising in the salivary glands.
In the lung, large cell carcinoma with neuroendocrine features is not readily recognizable as neuroendocrine by light microscopy, but it shows neuroendocrine differentiation by immunohisto-chemical staining and/or ultrastructural analysis (1). Although this type of tumor may be included within the category of undifferentiated carcinoma of the salivary glands, we failed to find such a case among those in our files.
In this study, we investigated alterations of several cell cycle regulators and tumor suppressor genes in parotid gland LCNECs. Generally, the overexpression of cyclins—positive growth regulators—may overwhelm the arrest mechanism of the normal cell cycle and lead to uncontrolled cell proliferation (19). Alternatively, the underexpression of negative growth regulators, such as the cyclin-dependent kinase inhibitors p16, p21Waf1, and p27Kip1, can have the same effect on the cell cycle (19, 20). For example, decreased or absent p27Kip1 expression has been implicated as a powerful negative prognostic factor in several malignancies (21, 22). In our cases, because immunohistochemistry revealed overexpression of cyclin D1 and reduced expression of p21Waf1 and p27Kip1, it is possible that these cell cycle regulators may play an integral part of carcinogenesis in parotid gland LCNECs. Deletions or mutations of p16 have a pivotal role in the development or progression of human cancer (23). Although LOH of the IFNA gene, flanking the p16 locus on chromosome 9p21, was seen in both of our cases, immunohistochemistry showed strong nuclear as well as cytoplasmic p16 reactivity in almost all of the tumor cells. Therefore, inactivation of p16 function by mutation, hypermethylation, or homozygous deletion may not contribute to tumorigenesis in our cases. In the lung, p53 gene alterations occur frequently in LCNEC and small cell carcinoma but are infrequent in typical and atypical carcinoids (24, 25). Our cases also showed p53 gene alterations as well as overexpression. Because our cases carried K-ras wild-type sequences, this oncogene may not be involved in the tumorigenesis of LCNEC of parotid gland origin; this has been the finding in LCNEC of the lung and uterine cervix (24, 25, 26). On the basis of the findings that bcl-2 was highly expressed in neuroendocrine tumor cells in combined LCNEC of the lung, Jiang et al. (27) proposed that bcl-2 may regulate cellular neuroendocrine differentiation. Compatible results were obtained in our cases, which also showed diffuse bcl-2 expression. Therefore, it is tempting to hypothesize that the multiple molecular-gene alterations—for example, p53 LOH; mutation and overexpression; LOH on chromosome 9p21; and aberrant expression of the epidermal growth factor receptor, cyclin D1, p21Waf1, and p27Kip1—have a negative influence on the biologic behavior of LCNEC of the parotid gland. To confirm this, however, we need more cases so that we can perform representative clinical and genetic analysis.
Because parotid gland LCNEC is rare, its biologic behaviors are unclear. In one of the previously reported cases, the patient was alive and well 3 years after parotidectomy and postoperative radiation therapy (9). Our two patients died of disease recurrence 5 months and 4 years after surgery, respectively. In the lung (2, 3) and uterine cervix (4), LCNECs have an unfavorable outcome, similar to that of small cell carcinomas. Gnepp and Wick (16) found that the 2- and 5-year survival rates for patients with small cell carcinomas arising in the major salivary glands were 70% and 46%, respectively. Generally, the prognosis of salivary gland small cell carcinomas seems to be more favorable than that of small cell carcinomas of bronchogenic or laryngeal origin, despite their high-grade malignancy (18). Chan et al. (28) suggested that at least some salivary gland small cell carcinomas may have a close biologic relationship or be identical to Merkel cell carcinoma based on their data indicating frequent CK20 immunoreactivity. Our LCNEC cases should be distinguished from Merkel cell carcinoma, which shows constant CK20 immunoreactivity. Negative immunoreactivity for CK20 in our cases, exhibiting highly aggressive biologic behavior, helps to exclude the possibility of Merkel cell carcinoma. Because salivary gland LCNEC is rare and its treatment varies, a definitive assessment of the prognosis is difficult. Further studies of more cases are needed to clarify the biologic behavior and suitable treatment of this rare neoplasm.
References
Travis WD, Linnoila RI, Tsokos MG, Hitchcock CL, Cutler GB Jr, Nieman L, et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma: an ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol 1991; 15: 529–553.
Dresler CM, Ritter JH, Patterson GA, Ross E, Bailey MS, Wick MR . Clinical-pathologic analysis of 40 patients with large cell neuroendocrine carcinoma of lung. Ann Thorac Surg 1997; 63: 180–185.
Travis WD, Rush W, Flieder DB, Falk R, Fleming MV, Gal AA, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol 1998; 22: 934–944.
Gilks CB, Young RH, Gersell DJ, Clement PB . Large cell neuroendocrine [corrected] carcinoma of the uterine cervix: a clinicopathologic study of 12 cases. Am J Surg Pathol 1997; 21: 905–914.
Chetty R, Batitang S, Govender D . Large cell neuroendocrine carcinoma of the thymus. Histopathology 1997; 31: 274–276.
Matsui K, Jin XM, Kitagawa M, Miwa A . Clinicopathologic features of neuroendocrine carcinomas of the stomach: appraisal of small cell and large cell variants. Arch Pathol Lab Med 1998; 122: 1010–1017.
Hailemariam S, Gaspert A, Komminoth P, Tamboli P, Amin M . Primary, pure, large-cell neuroendocrine carcinoma of the urinary bladder. Mod Pathol 1998; 11: 1016–1020.
Hui KK, Luna MA, Batsakis JG, Ordonez NG, Weber R . Undifferentiated carcinomas of the major salivary glands. Oral Surg Oral Med Oral Pathol 1990; 69: 76–83.
Larsson LG, Donner LR . Large cell neuroendocrine carcinoma of the parotid gland: fine needle aspiration, and light microscopic and ultrastructural study. Acta Cytol 1999; 43: 534–536.
Nagao K, Matsuzaki O, Saiga H, Sugano I, Shigematsu H, Kaneko T, et al. Histopathologic studies of undifferentiated carcinoma of the parotid gland. Cancer 1982; 50: 1572–1579.
Cawkwell L, Bell SM, Lewis FA, Dixon MF, Taylor GR, Quirke P . Rapid detection of allele loss in colorectal tumours using microsatellites and fluorescent DNA technology. Br J Cancer 1993; 67: 1262–1267.
Nagao T, Ishida Y, Sugano I, Tajima Y, Matsuzaki O, Hino T, et al. Epstein-Barr virus-associated undifferentiated carcinoma with lymphoid stroma of the salivary gland in Japanese patients: comparison with benign lymphoepithelial lesion. Cancer 1996; 78: 695–703.
Huntrakoon M . Neuroendocrine carcinoma of the parotid gland: a report of two cases with ultrastructural and immunohistochemical studies. Hum Pathol 1987; 18: 1212–1217.
Scher RL, Feldman PS, Levine PA . Small-cell carcinoma of the parotid gland with neuroendocrine features. Arch Otolaryngol Head Neck Surg 1988; 114: 319–321.
Mair S, Phillips JI, Cohen R . Small cell undifferentiated carcinoma of the parotid gland: cytologic, histologic, immunohistochemical and ultrastructural features of a neuroendocrine variant. Acta Cytol 1989; 33: 164–168.
Gnepp DR, Wick MR . Small cell carcinoma of the major salivary glands: an immunohistochemical study. Cancer 1990; 66: 185–192.
Seifert G, Sobin L . Histological typing of salivary gland tumors (World Health Organization). 2nd ed.New York:Springer-Verlag; 1991.
Ellis GL, Auclair PL . Tumors of the Salivary Glands. Atlas of Tumor Pathology. Washington, DC:Armed Forces Institute of Pathology; 1996.
Hall M, Peter G . Genetic alterations of cyclins, cyclin-dependent kinases, and cdk inhibitors in human cancers. Adv Cancer Res 1996; 68: 67–108.
Elledge SJ, Winston J, Harper JW . A question of balance: the role of cyclin-kinase inhibitors in development and tumorigenesis. Trends Cell Biol 1996; 6: 388–393.
Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, et al. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med 1997; 3: 227–230.
Tsihlias J, Kapusta LR, DeBoer G, Morava-Protzner I, Zbieranowski I, Bhattacharya N, et al. Loss of cyclin-dependent kinase inhibitor p27Kip1 is a novel prognostic factor in localized human prostate adenocarcinoma. Cancer Res 1998; 58: 542–548.
Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA . Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994; 368: 753–756.
Przygodzki RM, Finkelstein SD, Langer JC, Swalsky PA, Fishback N, Bakker A, et al. Analysis of p53, K-ras-2, and C-raf-1 in pulmonary neuroendocrine tumors: correlation with histological subtype and clinical outcome. Am J Pathol 1996; 148: 1531–1541.
Onuki N, Wistuba II, Travis WD, Virmani AK, Yashima K, Brambilla E, et al. Genetic changes in the spectrum of neuroendocrine lung tumors. Cancer 1999; 85: 600–607.
Wistuba II, Thomas B, Behrens C, Onuki N, Lindberg G, Albores-Saavedra J, et al. Molecular abnormalities associated with endocrine tumors of the uterine cervix. Gynecol Oncol 1999; 72: 3–9.
Jiang SX, Kameya T, Shoji M, Shinada J, Yoshimura H . The significance of frequent and independent p53 and bcl-2 expression in large-cell neuroendocrine carcinomas of the lung. Mod Pathol 1999; 12: 362–369.
Chan JKC, Suster S, Wenig BM, Tsang WYW, Chan JBK, Lau ALW . Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol 1997; 21: 226–234.
Acknowledgements
The authors thank Kenichi Hasegawa and Atsufumi Ohba, Department of Surgical Pathology, Teikyo University, School of Medicine, Ichihara Hospital, Japan, for the excellent photographs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nagao, T., Sugano, I., Ishida, Y. et al. Primary Large-Cell Neuroendocrine Carcinoma of the Parotid Gland: Immunohistochemical and Molecular Analysis of Two Cases. Mod Pathol 13, 554–561 (2000). https://doi.org/10.1038/modpathol.3880096
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880096
Keywords
This article is cited by
-
Primary Large Cell Neuroendocrine Carcinoma of the Parotid Gland. Report of a Rare Case
Head and Neck Pathology (2021)
-
P53 Gene Mutation Identified by Next Generation Sequencing in Poorly Differentiated Neuroendocrine Carcinoma of the Nasal Cavity
Head and Neck Pathology (2019)
-
NUT Carcinoma of the Sublingual Gland
Head and Neck Pathology (2016)
-
Neuroendokrine epitheliale Tumoren des oberen Respirationstrakts
Der Pathologe (2015)
-
Large cell neuroendocrine carcinoma of the head and neck: a distinct clinicopathologic entity
European Archives of Oto-Rhino-Laryngology (2014)
