
Abstract
Few reported cases of gliosarcomas or glioblastomas with epithelial-like areas exist. Most cases were originally diagnosed as metastatic carcinoma. Focal expression of glial fibrillary acidic protein has helped characterize these tumors as having a glial origin. We report a case of gliosarcoma with multifocal, extensive areas of well-differentiated carcinoma; demonstrating squamous and glandular differentiation. The expression of glial fibrillary acidic protein and epithelial phenotype were mutually exclusive. We performed extensive immunohistochemical analyses and comparative genotypic analysis using microdissection to secure representative glial and epithelial components. Loss of heterozygosity was analyzed with a panel of 12 polymorphic microsatellite markers designed to indicate allelic loss and situated in proximity to known tumor suppressor genes located on chromosomes 1p, 9p, 10q, 17p and 19q. We found comparable patterns of acquired allelic loss between the glial and carcinomatous components, strongly supporting the monoclonal origin of this neoplasm. This case represents an extreme form of phenotypic divergence in a malignant glioma, and constitutes a difficult diagnostic challenge. This heterogeneity reflects the potential for a range of phenotypic expression in malignant gliomas that needs to be recognized. We suggest microdissection genotyping as a molecular technique to better characterize these tumors.
Similar content being viewed by others

Main
Glioblastoma multiforme (glioblastoma) is the most common malignant brain tumor in adults comprising 12–15% of adult intracranial neoplasms and 50–60% of astrocytic neoplasms.1 This tumor can have heterogeneous cellular composition including small, lipidized, undifferentiated, granular, giant cells, and bipolar fusiform cells, hence the name glioblastoma multiforme. Gliosarcomas are defined as glioblastoma variants with a biphasic tissue pattern of glial and mesenchymal differentiation, and they are reported to share significant clinical and genetic similarities.2 The sarcomatous areas commonly resemble fibrosarcoma, but may show a variety of lines of mesenchymal differentiation, such as bone and cartilage3 or muscle.4, 5 Rare reports have noted distinctive epithelial histology within glioblastoma or gliosarcomas displaying squamoid or glandular appearances with negative immunohistochemical staining for glial fibrillary acidic protein (GFAP) and positivity for cytokeratin expression.6, 7, 8, 9, 10 These unusual cases can pose not only diagnostic dilemmas, but clinical management issues as to whether these distinctive areas represent metastasis from an extracranial tumor, as has been described,11 or a primary manifestation of the high-grade astrocytic neoplasm. Oh and Prayson12 has suggested the use of GFAP and CAM 5.2 immunostains in order to differentiate metastatic carcinoma from glioblastoma.
Primary and secondary glioblastoma derived from differentiated astrocytes or precursor cells follow distinctly different molecular genetic pathways. Epidermal growth factor receptor (EGFR) and MDM2 (mouse double minute 2; p53 binding and inactivation protein) amplification and overexpression are a feature early in the evolution of approximately one-half of primary glioblastomas, while p53 mutations, overexpression and amplification of platelet-derived growth factor receptor alpha (PDGFR-α), and loss of heterozygosity for 19q are early features of secondary glioblastoma.13, 14, 15, 16 Gliosarcomas have a distinct genetic profile, similar to glioblastomas except for the amplification of EGFR,17 and recent studies suggest that amplification of genes on proximal 12q could facilitate the development of a sarcomatous genotype.18 To date, little is known about the molecular genetics of epithelial elements seen in conjunction with glioblastoma or gliosarcomas. A recent study reports the same pattern of TP53 mutations in astrocytic areas and in areas of epithelial differentiation.19 This same finding had been originally described for glial and mesenchymal areas in gliosarcomas.20 We report here the histologic and genotypic analysis of a gliosarcoma with an extensive carcinomatous component.
Case report
A 66-year-old Caucasian man developed chronic headaches unresponsive to over-the-counter analgesics approximately 1 month prior to presentation. Concomitant symptoms included forgetfulness, confusion, and sporadic vomiting. A magnetic resonance image of the brain revealed a 6 × 5 × 4.5 cm solid, multicystic, and multilobulated right frontal lobe mass involving the genu of the corpus callosum and septum pellucidum with associated surrounding vasogenic edema with contrast enhancement and midline shift (Figure 1). The differential diagnoses based on the radiologic findings included a high-grade glioma, lymphoma, or metastases. A metastatic work-up was negative. The patient underwent a right craniotomy and resection of the right frontal mass.

Contrast-enhanced MRI showing a 6.4 cm partially cystic and solid mass in the right frontal lobe involving the genu of the corpus callosum with subfalcine herniation. Enhancement of some central solid areas with peripheral enhancement of the cystic region is seen.
Materials and methods
Histology
Sections of tumor material submitted were fixed in 10% buffered formalin overnight, processed and sectioned by routine histologic techniques after paraffin embedding. Sections were then stained with hematoxylin and eosin.
Immunophenotypic Characterization
Immunohistochemical analyses were performed on the formalin-fixed, paraffin-embedded tissue. Primary antibodies were directed against the following antigens: thyroid transcription factor (TTF), cytokeratin 7 (CK7), cytokeratin 20 (CK20), cytokeratin AE1/3, (Ki-67) proliferation index, p53 tumor suppressor protein, GFAP, all muscle actin (HHF35), Melan A, alpha fetoprotein (AFP), beta human chorionic gonadotropin (β-HCG) (all antibodies from DakoCytomation California, Inc., Carpinteria, CA, USA), placental alkaline phosphatase (PLAP) (Biogenex, Biogenex, San Ramon, CA, USA), synaptophysin (Ventana, Ventana Medical Systems, Inc., Tucson, AZ, USA), and CAM 5.2 (Becton, Dickinson and Company, San Jose, CA, USA). All immunostaining was performed on Dako or Ventana automated immunostainers. Mucicarmine stain was performed according to Mayer's method.
Microdissection Genotyping
Microdissection genotyping was performed on serial 4 μm thick histologic sections. Using the original hematoxylin–eosin-stained section as a guide, five separate targets were microdissected from each unstained slide using a hand-held scalpel under stereomicroscopic observation (Olympus SZ-40 stereomicroscope, Olympus USA, Inc. Melville, NY, USA). Samples were taken from areas demonstrating gliosarcoma, carcinoma, and areas with a mix of tumor and non-neoplastic tissue. Aliquots of each microdissected sample were PCR amplified as previously described.21 Each sample was aliquoted into 15 separate PCR reactions for individual polymorphic microsatellites situated at five genomic regions in proximity to known tumor suppressor genes known or highly suspected to undergo deletional damage in gliomas. The specific markers used with corresponding cytogenetic localization and GenBank accession information were 1p34:MYCL[M19720], 1p36:D1S407[L18040], 1p22:D1S1172[L17654], 1p36:D1S1193 [L30480], 9p21:D9S251[L18726], 9p23:D9S254[L18050], 10q23:D10S520[L16357], 10q23:D10S1173[L30341], 17p13:D17S974[G07961], 17p13:D17S1289[G09615], 19q:D19S400[L16430], 19q:D19S559[L30499]. PCR amplification was designed to generate an amplicon less than 200 bp long using synthetic oligonucleotide primers flanking each microsatellite. Oligonucleotide primers were created with 5′ fluorescent moieties (FAM, HEX, NED) suitable for fragment analysis. The PCR products were analyzed by capillary electrophoresis according to the manufacturer's instructions (ABI 3100, Applied Biosystems, Foster City, CA, USA). Allele peak heights and lengths were used to define the presence or absence of allelic imbalance for a given sample. Non-neoplastic microdissected tissue samples were first evaluated for informative status with respect to individual alleles. When a particular microsatellite marker in a normal tissue sample manifested only a single peak, the patient was designated as noninformative for that marker. For informative subjects with respect to a specific marker, alleles were assessed as being in balance when the ratio of the individual allele peaks fell within the range of 0.66–1.50. Values beyond this range were classified as being allelic imbalance within the following two categories. Low-level allelic imbalance was said to exist when the microsatellite allelic peak height ratios fell into the range 0.50–0.66 or 1.50–2.00. High-level allelic imbalance was present when the allele ratios fell below 0.50 or above 2.00.
Results
Morphologic and Immunophenotypic Characterization
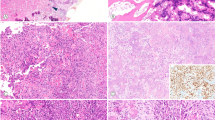
The craniotomy specimen consisted of multiple fragments of pale, tan and focally hemorrhagic soft tissue 2.5 × 1.5 × 1 cm in aggregate. The tumor was composed of malignant appearing glial cells demonstrating highly pleomorphic astrocytic nuclei, occasional multinucleated cells, and gemistocytes (Figure 2a) with large areas of necrosis adjacent to viable tumor (Figure 2a, inset). Mitotic figures and endothelial proliferation were easily identified (Figure 2b). Distinct areas of the tumor were composed of atypical spindled cells within a dense collagenous stroma (Figure 2c) with occasional atypical mitotic cells (arrow, inset, Figure 2c). Reticulin stain (not shown) of sarcoma-like areas shows numerous tightly packed interlacing fibers around atypical spindle cells. Other areas had chondroblastic-like cells (Figure 2d) with some bi- and multinucleated forms set in a chondroid matrix (arrows, Figure 2d). Sarcoma-like areas were seen encroaching and surrounding glial and epithelial areas. GFAP staining highlighted the astrocytic component of the neoplasm while not staining epithelial or sarcomatous elements (Figure 2e). Nests of atypical squamoid cells with focal individual cell keratinization and keratin pearls were interspersed but separate and distinct from the malignant glial cells (Figure 2f). The carcinomatous portion demonstrated focally trabecular and papillary growth patterns (Figure 2g). Vague ‘rosette’ like structures could be visualized in some of the carcinomatous areas showing a trabecular growth pattern (inset, Figure 2g) while other areas demonstrated glandular formation with mucin production (arrows, Figure 2h Distinct perivascular pseudorosetting, however, was not seen. Numerous mitoses were also seen within the carcinomatous areas. The malignant glial component was diffusely and strongly positive for GFAP. Some areas showed a more gradual transition from the malignant glial component to the more poorly differentiated carcinomatous areas best demonstrated on GFAP staining. Areas showing epithelial and glandular differentiation showed strong, diffuse staining for low molecular weight cytokeratin (CAM5.2) (Figure 3a) and cytokeratin 7 (CK7) (inset, Figure 3a), with focal positivity for cytokeratin 20 (CK20) (not shown). Tumor suppressor protein, p53, staining of the glial component demonstrated moderately numerous positive cells (Figure 3b) while epithelial carcinomatous elements showed an even greater number of positive cells (inset, Figure 3b). (Ki-67) proliferation index was approximately 30% within the malignant glial cells (Figure 3c) and <50% within the carcinomatous areas (inset, Figure 3c). Both tumor elements stained negatively for Melan A, PLAP, HHF35, β-HCG, AFP, and TTF.

Medium power view of the craniotomy specimen demonstrating highly cellular astrocytic neoplasm with pleomorphic astrocytes, multinucleated cells, and gemistocytes (a) with large areas of tumor necrosis adjacent to viable tumor (inset a). High power shows numerous mitoses and endothelial proliferation (b). Focal areas within the tumor were composed of atypical spindle cells within a collagenous stroma component (c) with occasional atypical mitotic figures (inset c). Some areas showed atypical cells within a distinctly chondroid matrix (arrows, d). GFAP stain highlights astrocytic component but not epithelial or sarcoma-like regions (medium power, e). At high power some carcinomatous areas exhibited squamoid appearing cells with keratin pearl formation and individual cell keratinization (f), focally trabecular and papillary growth patterns (g) and vague rosette formation (inset, g). Gland formation with intraluminal and intracellular mucin was seen by mucicarmine stain (arrows, h).

Carcinomatous areas stained strongly and diffusely positive for CAM 5.2 (a) and for CK7 (inset, a). Glial (b) and carcinomatous areas (inset, b) demonstrated staining for p53. Proliferation marker, (Ki-67), revealed numerous positive nuclei within glial elements (c) and more than 50% positivity within carcinoma areas (inset, c).
Genotyping Results
Results of microdissection genotyping are shown in Table 1. The tumor was microdissected at sites corresponding to gliosarcomatous and carcinomatous morphology. Microdissected tissue corresponding to a composite of non-neoplastic cellular elements and tumor was used to establish microsatellite marker informativeness. The presence of potentially mutated alleles in the latter did not interfere with determination of marker informativeness. A total of nine informative microsatellite markers were defined from the total panel of 12 markers. Using these informative markers the gliomatous regions of the tumor manifested five allelic loss alterations. All of these mutated markers as well as the specific alleles affected by loss were shown to be present in the mixed gliomatous and non-neoplastic growth areas. Three of the markers were concordantly affected in all microdissected tissue samples. This established the monoclonal character of the morphologically heterogeneous tumor in this instance.
Discussion
Gliosarcomas are rare biphasic neoplasms of the central nervous system composed of glioblastoma admixed with a sarcomatous component. Several studies agree with the fact that gliosarcomas and glioblastoma cannot be distinguished clinically.2, 22 Rarely, gliosarcomas with focal epithelial features have been described.6, 7, 8 The interpretation of these lesions as variants of glioblastoma have been based on partial expression of GFAP in the epithelial areas as well as no evidence of another primary tumor. To our knowledge, only the study of Mueller et al 19 investigated clonality in these tumors by studying the patterns of TP53 mutation in different areas of the tumor. The spreading of metastasis into cranial tumors has been reported.11 Therefore, the possibility of a collision tumor in these situations has to be considered and carefully evaluated. The case presented here is an extreme form of phenotype divergence in a malignant glioma and constitutes a difficult diagnostic challenge, given the fact that at least half of the tumor is composed of carcinoma-like areas. In fact, most of the reported cases of glioblastoma with epithelial differentiation, were originally diagnosed as metastatic tumors. This heterogeneity reflects the potential for a range of phenotypic expression in malignant gliomas that needs to be recognized.
During embryonic development, conversions from epithelium to mesenchyme, and vice versa, occur normally, being all part of the morphogenetic process and tissue remodeling.23 Indeed, there is evidence that this change occurs in human diseases, including the development of carcinosarcomas.24 Carcinosarcomas are malignant tumors having a mixture of carcinoma and sarcoma containing differentiated mesenchymal elements such as malignant cartilage, bone and muscle.25, 26 Recent data confirm that most, but not all, carcinosarcomas are monoclonal,27, 28, 29 and that the carcinomatous element is the driving force and the sarcomatous component is derived from the carcinoma or from a stem cell that undergoes divergent differentiation. It is however important to differentiate these cases from true collision tumors, since the prognosis will vary.
The relatedness between the morphologically distinct tumor growth patterns was evaluated in this case by a system of microdissection genotyping combining precise tissue microdissection followed by mutational profiling. This approach provides information not only on the status of allelic loss to a particular marker, but also which specific allele is affected by deletion when evidence for this change is present. As such it provides a detailed fingerprint with which to compare different regions of neoplastic growth from the standpoint of an acquired profile of allelic loss damage. The greater the degree of concordance of an allelic loss profile, the stronger the evidence is provided that the two neoplastic regions are closer to each other in tumorigenesis. In this case, the gliomatous growth pattern of tumor manifested five allelic loss alterations. Notably, all five of these specific allelic losses including the unique allele affected by deletion was demonstrable in the carcinomatous growth areas. This provides compelling evidence to support the contention that this is one neoplasm with divergent growth rather than independent primary tumors that secondarily have collided in location.
A limitation in this case study was the lack of completely non-neoplastic tissue as a control for mutational profiling. Surgical neuropathology is especially challenging in this regard since it is often the case that careful attention is paid during surgery to avoid the removal of histologically normal appearing brain parenchyma. This was the case in this patient. However, the issue was addressed by microdissection of peritumoral tissue representing a mixture of non-neoplastic and neoplastic cellular elements. This approach provided samples with sufficient non-neoplastic DNA upon which to definitely establish microsatellite marker informativeness.
In summary, we present the unique case of a gliosarcoma with a morphologically extensive carcinomatous component. We suggest microdissection genotyping as the technique of choice for characterizing these difficult diagnostic dilemmas.
References
Burger PC, Kleihues P . Cytologic composition of the untreated glioblastoma with implications for evaluation of needle biopsies. Cancer 1989;63:2014–2023.
Galanis E, Buckner JC, Dinapoli RP, et al. Clinical outcome of gliosarcoma compared with glioblastoma: North Central Cancer Treatment Group results. J Neurosurg 1998;89:425–430.
Tada T, Katsuyama T, Aoki T, et al. Mixed glioblastoma and sarcoma with osteoid-chondral tissue. Clin Neuropathol 1987;6:160–163.
Barnard RO, Bradford R, Scott T, et al. Gliomyosarcoma. Report of a case of rhabdomyosarcoma arising in a malignant glioma. Acta Neuropathol (Berl) 1986; 69:23–27.
Haddad SF, Moore SA, Schelper RL, et al. Smooth muscle can comprise the sarcomatous component of gliosarcomas. J Neuropathol Exp Neurol 1992;51:493–498.
Kato K, Watanabe M . Glioblastoma with epithelial appearance: a case report. Brain Tumor Pathol 1999; 16:45–48.
Kepes JJ, Fulling KH, Garcia JH . The clinical significance of ‘adenoid’ formations of neoplastic astrocytes, imitating metastatic carcinoma, in gliosarcomas. A review of five cases. Clin Neuropathol 1982;1:139–150.
Kim DS, Kang SK, Chi JG . Gliosarcoma: a case with unusual epithelial feature. J Korean Med Sci 1999; 14:345–350.
Moork SJ, Rubinstein LJ, Kepes JJ . Patterns of epithelial metaplasia in malignant gliomas. I. Papillary formations mimicking medulloepithelioma. J Neuropathol Exp Neurol 1988;47:93–100.
Mork SJ, Rubinstein LJ, Kepes JJ, et al. Patterns of epithelial metaplasia in malignant gliomas. II. Squamous differentiation of epithelial-like formations in gliosarcomas and glioblastomas. J Neuropathol Exp Neurol 1988;47:101–118.
Muller W, Schroder R . Spreading of metastases into cranial tumors: metastasis of a breast carcinoma to a pilocytic astrocytoma. Clin Neuropathol 1999;18:109–112.
Oh D, Prayson RA . Evaluation of epithelial and keratin markers in glioblastoma: an immunohistochemical study. Arch Pathol Lab Med 1999;123:917–920.
Fults D, Pedone C . Deletion mapping of the long arm of chromosome 10 in glioblastoma. Genes Chromosomes Cancer 1993;7:173–177.
Kleihues P, Ohgaki H . Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-oncol 1999;1:44–51.
von Deimling A, Nagel J, Bender B, et al. Deletion mapping of chromosome 19 in human gliomas. Int J Cancer 1994;57:676–680.
Watanabe K, Tachibana O, Sata K, et al. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 1996;6:217–223.
Reis RM, Konu-Lebleblicioglu D, Lopes JM, et al. Genetic profile of gliosarcomas. Am J Pathol 2000; 156:425–432.
Actor B, Cobbers JM, Buschges R, et al. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosomes Cancer 2002;34:416–427.
Mueller W, Lass U, Herms J, et al. Clonal analysis in glioblastoma with epithelial differentiation. Brain Pathol 2001;11:39–43.
Biernat W, Aguzzi A, Sure U, et al. Identical mutations of the p53 tumor suppressor gene in the gliomatous and the sarcomatous components of gliosarcomas suggest a common origin from glial cells. J Neuropathol Exp Neurol 1995;54:651–656.
Guo Z, Wu F, Asplund A, et al. Analysis of intratumoral heterogeneity of chromosome 3p deletions and genetic evidence of polyclonal origin of cervical squamous carcinoma. Mod Pathol 2001;14:54–61.
Lutterbach J, Guttenberger R, Pagenstecher A . Gliosarcoma: a clinical study. Radiother Oncol 2001;61:57–64.
Guarino M, Micheli P, Pallotti F, et al. Pathological relevance of epithelial and mesenchymal phenotype plasticity. Pathol Res Pract 1999;195:379–389.
Guarino M . Epithelial-to-mesenchymal change of differentiation. From embryogenetic mechanism to pathological patterns. Histol Histopathol 1995;10: 171–184.
Koss MN, Hochholzer L, Frommelt RA . Carcinosarcomas of the lung: a clinicopathologic study of 66 patients. Am J Surg Pathol 1999;23:1514–1526.
McCluggage WG . Malignant biphasic uterine tumours: carcinosarcomas or metaplastic carcinomas? J Clin Pathol 2002;55:321–325.
van den Berg W, Tascilar M, Offerhaus GJ, et al. Pancreatic mucinous cystic neoplasms with sarcomatous stroma molecular evidence for monoclonal origin with subsequent divergence of the epithelial and sarcomatous components. Mod Pathol 2000;13:86–91.
Watanabe M, Shimizu K, Kato H, et al. Carcinosarcoma of the uterus: immunohistochemical and genetic analysis of clonality of one case. Gynecol Oncol 2001;82:563–567.
Mukhopadhyay S, Shrimpton AE, Jones LA, et al. Carcinosarcoma of the urinary bladder following cyclophosphamide therapy: evidence for monoclonal origin and chromosome pp allelic loss. Arch Pathol Lab Med 2004;128:e8–e11.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ozolek, J., Finkelstein, S. & Couce, M. Gliosarcoma with epithelial differentiation: immunohistochemical and molecular characterization. A case report and review of the literature. Mod Pathol 17, 739–745 (2004). https://doi.org/10.1038/modpathol.3800109
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800109
