
Abstract
Study design:
Case report.
Objective:
To describe an unusual case of progressive pulmonary hypertension due to recurrent pulmonary embolism in a chronically paralyzed spinal cord injury patient.
Setting:
Veterans Administration Hospital, West Roxbury, MA, USA.
Subject:
A 57-year-old man, tetraplegic, sensory incomplete and motor complete for 30 years due to a diving accident, complained of lightheadedness and shortness of breath intermittently for 7 years. Examination during the latest episode revealed anxiety, confusion, respirations 28 per min, blood pressure 80/60 mmHg, and arterial pH 7.41, 28 mmHg, 95 mmHg on 2 l of oxygen. A chest film 2 weeks earlier had revealed a right-sided cutoff of pulmonary vasculature; the current film showed right-sided pleural effusion. Review of EKGs showed a trend of increasing right axis deviation with recovery and recurrences during the previous 9 years and a current incomplete right bundle branch block with clockwise rotation and inverted T waves in V1–4. Computerized tomography with contrast material revealed small pulmonary emboli, but only in retrospect. The patient died shortly after scanning.
Autopsy:
The pulmonary arteries were free of thromboemboli on gross examination but medium and small-sized arteries were constricted or obliterated with thrombotic material microscopically. The estimated ages of the thromboemboli ranged from days to years. The right ventricle was hypertrophied; the coronary arteries were patent.
Conclusion:
Recurrent pulmonary emboli resulted in chronic pulmonary hypertension and eventual death in a patient with chronic tetraplegia.
Similar content being viewed by others
Introduction
In spite of the relatively high prevalence of fatal pulmonary embolism in acute spinal cord injury (SCI),1, 2, 3 fatal PE in the chronic SCI patient is seldom reported.4, 5, 6 Recurrent PE in the chronic SCI patient, resulting in chronic and eventually fatal pulmonary hypertension, is rarer still. Considering the possibility that such cases are overlooked, this paper describes the first report.
Case report
A 57-year-old man, tetraplegic at C5, sensory incomplete and motor complete for 30 years after a diving accident, was hospitalized for shortness of breath and abdominal discomfort. He had sustained brief episodes of dyspnea and light headedness on several occasions during the previous 7 years. Vital capacity was 1.6 l (average for able-bodied at his age 4.9 l) and forced expiratory volume at 1 s was 94% on testing 5 years before this admission. A sleep study had not been carried out. There was a past history of recurrent urinary tract infections, urethral stricture, epididymitis, cholecystectomy, bowel distention and severe, recurrent spasticity. Urinary drainage was by indwelling catheter.
Initially treated for a urinary tract infection, he developed diarrhea, a tympanitic abdomen, and large bowel ileus by abdominal radiograph. Stool became positive for C. difficile toxin.
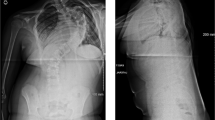
An episode of shortness of breath and slurred speech developed. Exam revealed T 95, P 72 R 28 BP 80/60 and 88% oxygen saturation on room air. Blood pressures had ranged from 72/66 to 140/80, median 91/63 for 14 readings in the 7 years prior to the current admission. Arterial blood pH was 7.41, 28, 95 mmHg on 2 l of oxygen. The hematocrit was 43%, white count 11 000/m3. Coagulation studies were not carried out. The serum sodium was 121, chloride 95, and bicarbonate 17 mEq/l. (Baseline sodium and bicarbonate values were 132 to 142, median 138 mEq/l, and 24–29, median 26 mEq/l for seven samples over 5 years, ending 2 years before this admission.) A chest film 2 weeks earlier had revealed a cutoff of pulmonary vasculature on the right side (Figure 1). The current film showed right-sided pleural effusion. EKG showed right axis deviation, a new incomplete right bundle branch block (IRBBB), clockwise rotation, and right heart strain with inverted T waves in V1–V4 (Figure 2). A myocardial infarction was ruled out as the troponin and creatine kinase levels were normal. Computerized tomography (CT) with intravenous contrast revealed subsegmental emboli – but only in retrospect – and fluid in the pleural spaces. An echocardiogram was not performed.

Chest radiograph showing a cutoff of a segmental right pulmonary (arrow)

Electrocardiogram the day prior to the patient's death showing right axis deviation (+135°), right heart strain (T inverted in V1–V4), clockwise rotation, and incomplete right bundle branch block
Hypovolemia secondary to fluid loss from colitis was suspected. Intravenous saline and oral metronidazole were administered.
Subsequently, the patient became agitated and confused. Although oxygen saturation remained at 92% on supplemental oxygen, blood pressure was low and fluctuating. The patient then lost consciousness and blood pressure was unobtainable. No end of life decision having been made by the patient, cardiopulmonary resuscitation was attempted. This effort was unsuccessful, and the patient expired.
Autopsy report
Autopsy revealed that the heart weighed 350 g (normal 280–350 g), with right ventricular hypertrophy and a maximum thickness of 6 versus 12 mm for the left ventricle (normal ratio 1:3). The coronary arteries were patent, and there was no infarction of the left ventricle. Microscopic examination revealed fiber hypertrophy of the right ventricular muscle but no incipient necrotic or fibrotic changes.
The pleural cavities contained clear, serous fluid – 500 ml on the right and 250 ml on the left. The pleural surfaces focally showed hemorrhagic, fibrinous, and acute inflammatory exudate in some areas with reddened, firm areas in the underlying lung, suggestive of pulmonary infarcts. The lungs weighed 300 and 250 g on the right and left, respectively, with minimal atelectasis, and there was no significant evidence of emphysema. Microscopic examination revealed evidence of extensive thromboembolic change in medium- and small-sized pulmonary arteries throughout both lungs and scattered foci of chronic inflammation, interpreted as pulmonary infarctions.7 Recent, hyalinized thrombotic material and old, recanalized thrombi with the formation of multiple small vascular channels within the original inner elastic membrane of the involved artery were found (Figure 3). This pathology was seen in all sections taken from the lung and had resulted in marked narrowing of the lumen of the involved vessels, which varied from about 40% of the original bore up to virtual occlusion. In some instances there was formation of a secondary inner elastic membrane about the newly formed channels within the affected arteries. Indeed, several vessels showed evidence of recanalized thrombi within the new channels, indicating superimposed thromboembolic events. The distinction between intravascular thrombosis and pulmonary thromboembolism not being possible by histology alone, clinical correlates were enlisted for interpretation.

Top panel: pulmonary artery showing a recent intraluminal thrombus (upper arrow). There is concentric intimal hyperplasia inside normal medial muscle and narrowing by endothial ‘cushions’ (asterisks), formed from one or more old organized thrombi and an overlying organizing thrombus of more recent date (lower arrow). Stain: hematoxylin and eosin. Bottom panel: pulmonary artery occlusion by mature fibrous tissue, pierced by multiple recanalized vascular channels. Original lumen is demarcated by the inner elastic membrane (arrow). Stain: Van Gieson
The pelvic veins showed no naked-eye evidence of thrombosis, and the deep venous system of the legs was not dissected.
The thyroid gland was tan in cut section and weighed 10 g (normal 30 g). Microscopically the gland showed nodular architecture, atrophic follicles, interstitial fibrosis, and focal areas of chronic inflammation. Serum thyroid stimulating hormone (TSH), which became available after death, was 10.4 IU/ml (normal <5). A TSH level had been normal 2 years earlier.
The peritoneal cavity contained 500 ml of nonpurulent fluid. The bladder mucosa was hemorrhagic and the histology showed evidence of severe chronic focal inflammation in the muscularis and serosa. The kidneys were focally scarred with a few cysts and showed chronic pyelonephritis, microscopically. The prostate gland had a hypertrophied median lobe that impinged on the bladder neck. Microscopically, focal, infiltrating adenocarcinoma with perineural invasion, and high-grade intraepithelial neoplasia were found. A year before the prostate-specific antigen level had been 2.2 ng/ml (normal <4.0).
Comments
Pulmonary hypertension and pulmonary emboli
The clinical presentation of PE was suggested by the patient's recurrent dyspnea, anxiety, and confusion. Although these symptoms are nonspecific, they can represent poor cerebral perfusion and portend a poor prognosis in the setting of PE.8 The hypotension and hypoxemia were classical presentations for PE. The new onset of hypocarbia resulting from hyperventilation, is another possible manifestation of PE.9 Also, hyponatremia, representing high antidiuretic hormone levels (ADH) may have been induced by the hypotension of PE. Hypotension is a powerful stimulus to ADH secretion and can over-ride osmolality as a control for its secretion, particularly in tetraplegic patients.10, 11, 12 The onset of IRBBB by EKG is a recognized sign of PE. Review of previous EKG tracings revealed right axis deviation 9 years and 1 year before, suggesting previous showers of PE (Figure 4). Based on the symptoms and the EKG trends, the course of PE in this patient was chronically recurrent, terminating in a potentially recognizable picture of acute PE. Confirmation by CT was initially overlooked, partly due to the relative insensitivity of the technique to subsegmental emboli. Larger thromboemboli can fragment and migrate distally before testing is carried out. Pulmonary arteriography, a more sensitive test and the gold standard for detection of PE, might have imaged these small fragments more clearly.13 In this case, PE was confirmed only at autopsy, which demonstrated microscopic old and new thromboemboli and scattered gross and microscopic pulmonary infarctions.

The changing direction of the axis of the QRS complex on EKG tracings taken serially over a 10-year period is plotted against time. A right axis trend had developed at the start of this review period, slowly returned toward the initial value, then recurred during the last year
Autopsy revealed right ventricular hypertrophy. The normal weight of the lungs, and the absence of microscopic evidence of chronic passive congestion, indicated that chronic left ventricular failure was not the cause for chronic right ventricular strain. A negative work-up for acute myocardial infarction (normal troponin and CK enzyme levels) and the absence of coronary artery disease and myocardial fiber damage at autopsy ruled out ischemia as the cause of the EKG changes – bundle branch block and precordial T-wave inversion. Restrictive lung disease, evidenced by a reduced vital capacity without prolongation of forced expiration, is common in SCI patients,14 but has not been associated with pulmonary hypertension and right ventricular hypertrophy in this population. Sleep apnea is common in the tetraplegic patient15 and conceptually induces pulmonary hypertension intermittently during sleep due to hypoxemia. It is noteworthy that, in pulmonary hypertension of the able-bodied, sleep apnea is uncommon while pulmonary embolism can account for half of such cases.16, 17, 18
Fatal pulmonary embolism is not often reported in patients chronically paralyzed due to SCI. Six case reports have been found in the literature.4, 5, 6 These have all been diagnosed after an acute event. In contrast, the patient described here presented with recurrent, accumulated PE events, and ultimately fatal pulmonary hypertension. This course is suggested by the history, the serial analysis of EKG tracings, and the histopathology in this patient. A small percentage of chronically paralyzed patients do, in fact, develop clinically detectable deep venous thrombosis, so that fatal PE remains a threat, though the risk is poorly defined.19 The challenge of diagnosing PE in the chronically paralyzed SCI patient is apparent. Autopsy surveys of fatal PE in the able-bodied show that the majority of cases are clinically undiagnosed.20
Hypothyroidism
Thyroid disease is more common in the SCI population than in the able-bodied by an approximate factor of 10.21, 22 The small thyroid gland and moderate elevation of TSH support the diagnosis of hypothyroidism. The patient's low serum albumin and pleural and pelvic fluid collections are consistent with a vascular leak characteristic of this condition.23 Hypothyroidism also induces hyponatremia24 and therefore is an alternate explanation for hyponatremia in this patient. Finally, a case of PE and hypothyroidism has been reported.25 Thus thyroid disease is another complication to be aware of in the chronically paralyzed.
Cancer of the prostate gland
Cancer arising from the prostate is uncommon among spinal cord injury patients.26 A low prevalence is attributable to those with complete paralysis.27, 28, 29 This protection – possibly by interruption of the sympathetic neuroendocrine cycle controlling androgen production – is incomplete, however, with incomplete spinal cord injuries as in the patient presented.
Carcinoma of any size arising from any site can predispose to thromboembolic events.30, 31 In this patient, however, the prostate-specific antigen was within normal range the year prior to his death. In another possible link, the prostate was impinging on the bladder neck, creating an obstruction to urinary drainage and leading to hemorrhagic cystitis. However, the cystic veins were not examined for a source of pulmonary emboli.
Conclusion
A patient with long-standing tetraplegia developed chronic, recurrent pulmonary thromboembolism resulting in pulmonary hypertension, which was eventually fatal. The patient had also sustained chronic hemorrhagic cystitis, C. difficile colitis, hypothyroidism, and cancer of the prostate gland; but the role of these diseases in the death of this patient was uncertain.
The events of the case suggest that pulmonary embolism should be included in the differential diagnosis of dyspnea in not only the acute but the chronically paralyzed as well. Evidence for pulmonary hypertension by physical exam, chest films, and review of EKG tracings should be sought. Echocardiogram might confirm right ventricular dilation and pulmonary hypertension. Demonstration of an acute PE by CT scanning or by perfusion lung scanning may depend upon the timing of these tests and the rate of fragmentation and distal migration of the thromboembolus. A search for deep venous thrombosis by venous Doppler studies is probably useful in the presumption of PE as the cause of pulmonary hypertension. The prevalence of pulmonary hypertension due to pulmonary embolism in the SCI population is not known.
Whether anticoagulation is effective in the treatment of recurrent thromboemboli is undetermined.32
References
Tribe CR . Causes of death in early and late paraplegia. Paraplegia 1963; 1: 10–45.
Wolman L . The disturbance of circulation in traumatic paraplegia in acute and late stages: a pathological study. Paraplegia 1964; 2: 213–226.
DeVivo MJ, Kartus PL, Stover SL, Rutt RD, Fine PR . Cause of death for patients with spinal cord injuries. Arch Intern Med 1989; 149: 1761–1766.
Rossier VA, Brunner U . Sur initialen Behandlung der frischen traumtischen Quirschmittslasion. Schweiz Med Wschr 1964; 94: 362–370.
Freed MM, Bakst HJ, Barrie DL . Life expectancy, survival rates, and causes of death in civilian patients with spinal cord trauma. Arch Phys Med Rehabil 1966; 47: 457–463.
Nyquist RH, Bors E . Mortality and survival in traumatic myelopathy during nineteen years, from 1946 to 1965. Paraplegia 1976; 5: 22–48.
Garcia JG, Perlman MB, Ferro TJ, Johnson A, Jubiz W, Malik AB . Inflammatory events after fibrin microembolization. Alterations in alveolar macrophage and neutrophil function. Am Rev Respir Dis 1988; 137: 630–635.
Frisbie JH, Sharma GVRK . Pulmonary embolism manifesting as acute disturbances of behavior in patients with spinal cord injury. Paraplegia 1994; 32: 570–572.
Santolicandro A et al. Mechanisms of hypoxemia and hypocapnia in pulmonary embolism. Am J Respir Crit Care Med 1995; 152: 336–347.
Frisbie JH, Steele DJR . Postural hypotension and abnormalities of salt and water metabolism in myelopathy patients. Spinal Cord 1997; 35: 303–307.
Robertson GL, Shelton RL, Athar S . The osmoregulation of vasopressin. Kidney Int 1976; 10: 25–37.
Sved AF, McDowell FH, Blessing WW . Release of antidiuretic hormone in quadriplegic subjects in response to head-up tilt. Neurology 1985; 35: 78–82.
Winer-Muran HT et al. Suspected acute pulmonary embolism: evaluation with multi-detector row CT versus digital subtraction pulmonary arteriography. Radiology 2004; 233: 806–815.
Linn WS, Adkins RH, Gong H, Waters RL . Pulmonary function in chronic spinal cord injury: a cross-sectional survey of 222 southern California adult outpatients. Arch Phys Med Rehabil 2000; 81: 757–763.
Stockhammer E et al. Characteristics of sleep apnea syndrome in tetraplegic patients. Spinal Cord 2002; 40: 286–294.
Atwood Jr CW, McCrory D, Garcia JG, Abman SH, Ahearn GS . Pulmonary artery hypertension and sleep-disordered breathing: ACCP evidence-based clinical practice guidelines. Chest 2004; 126(Suppl 1): 72–77.
Runo JR, Loyd JE . Primary pulmonary hypertension. Lancet 2003; 361: 1533–1543.
Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL . Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984; 70: 580–587.
Kim SW et al. Prevalence of deep venous thrombosis in patients with chronic spinal cord injury. Arch Phys Med Rehabil 1994; 75: 965–968.
Lau G . Pulmonary thromboembolism is not uncommon – results and implications of a five-year study of 116 necropsies. Ann Acad Med Singapore 1995; 24: 356–365.
Prakash V, Lin MS, Song CH, Perkash I . Thyroid hypofunction in spinal cord injury patients. Paraplegia 1980; 18: 56–63.
Frisbie JH, Fisch B . Graves' disease complicating myelopathy. Paraplegia 1992; 30: 593–597.
Skelton CL, Sonnenblick EH . The cardiovascular system. In: Ingbar SH, Braverman LE (eds). The Thyroid 5th edn. JB Lippincott: Philadelphia 1986, p 1143.
Kleeman CR, Danovitch GM . The kidneys and electrolyte metabolism. In: Ingbar SH, Braverman LE (eds). The Thyroid 5th edn. JB Lippincott: Philadelphia 1986, pp 1152–1153.
Kollef MH . Reversible hypotension following pulmonary embolism in a 33-year-old woman with hypothyroidism and vitiligo. Chest 1993; 104: 1889–1891.
Ito TY, Mehta M . Adenocarcinoma of the prostate in paraplegics. Paraplegia 1976; 14: 101–104.
Frisbie JH, Binard J . Low prevalence of prostatic cancer among myelopathy patients. J Am Paraplegia Soc 1994; 17: 148–149.
Frisbie JH . Cancer of the prostate in myelopathy patients: lower risk in higher levels of paralysis. J Spinal Cord Med 2001; 24: 92–95.
Favazza T, Lyon W, Midha M, Grob BM, Nseyo UO . A lower prevalence of prostate cancer in patients with tetraplegia. J Spinal Cord Med 2003; 26: 286 (Abstract).
Piccioli A, Prandoni P . Idiopathic venous thromboembolism as a first manifestation of cancer. Haemostasis 2001; 31(Suppl 1): 37–39.
Haim N, Lanir N, Hoffman R, Haim A, Tsalik M, Brenner B . Acquired activated protein C resistance is common in cancer patients and is associated with venous thromboembolism. Am J Med 2001; 110: 91–96.
Cundiff DK . Does anticoagulant treatment reduce the mortality of acute pulmonary embolism? Arch Intern Med 2001; 161: 2148.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Frisbie, J., Sharma, G., Brahma, P. et al. Recurrent pulmonary embolism and pulmonary hypertension in chronic tetraplegia. Spinal Cord 43, 625–630 (2005). https://doi.org/10.1038/sj.sc.3101745
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.sc.3101745
