
Abstract
Elevated platelet serotonin (5-hydroxytryptamine, 5-HT) is found in a subset of children with autism and in some of their first-degree relatives. Indices of the platelet serotonin system, including whole blood 5-HT, 5-HT binding affinity for the serotonin transporter (Km), 5-HT uptake (Vmax), and lysergic acid diethylamide (LSD) receptor binding, were previously studied in 24 first-degree relatives of probands with autism, half of whom were selected for elevated whole blood 5-HT levels. All subjects were then genotyped for selected polymorphisms at the SLC6A4, HTR7, HTR2A, ITGB3, and TPH1 loci. Previous studies allowed an a priori prediction of SLC6A4 haplotypes that separated the subjects into three groups that showed significantly different 5-HT binding affinity (Km, p=0.005) and 5-HT uptake rate (Vmax, p=0.046). Genotypes at four individual polymorphisms in SLC6A4 were not associated with platelet 5-HT indices. Haplotypes at SLC6A4 and individual genotypes of polymorphisms at SLC6A4, HTR7, HTR2A, ITGB3, and TPH1 showed no significant association with whole blood 5-HT. Haplotype analysis of two polymorphisms in TPH1 revealed a nominally significant association with whole blood 5-HT (p=0.046). These initial studies of indices of the 5-HT system with several single-nucleotide polymorphisms at loci in this system generate hypotheses for testing in other samples.
Similar content being viewed by others

INTRODUCTION
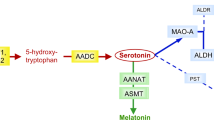
Twin studies suggest that autism has a complex genetic etiology (Folstein and Rutter, 1977; Bailey et al, 1995). One clue into the pathophysiology of autism is elevated whole blood serotonin (5-hydroxytryptamine, 5-HT), present in approximately one-third of patients (Schain and Freedman, 1961; Cook and Leventhal, 1996). Hyperserotonemia has been found to be familial and to be associated with recurrence risk of autism within families (Abramson et al, 1989; Cook et al, 1990; Piven et al, 1991). Thus, genetic variation in the 5-HT pathway is likely to shed light on the genetic liability to the syndrome.
In our previous study of first-degree relatives of autism probands, whole blood 5-HT levels were found to correlate with 5-HT transport uptake Vmax. Additionally, the hyperserotonemic subgroup was found to have decreased [3H]-lysergic acid diethylamide (LSD)-labeled receptor binding. These two findings clustered in distinct subgroups. Of the eight hyperserotonemic subjects who had both 5-HT uptake and LSD binding studies performed, four subjects had 5-HT uptake that was at least 1 SD above the highest normoserotonemic subject and three different subjects had LSD binding that was at least 0.95 SDs below the lowest normoserotonemic subject (Cook et al, 1993).
The correlation between 5-HT transport and whole blood 5-HT could arise from variation in the gene encoding the 5-HT transporter (SERT, SLC6A4). Linkage and association studies have implicated SLC6A4 in autism (Yonan et al, 2003; Stone et al, 2004; Devlin et al, 2005; Sutcliffe et al, 2005). The SLC6A4 promoter polymorphism HTTLPR long–long (L/L) genotype is associated with increased 5-HT uptake into the platelet (Greenberg et al, 1999; Anderson et al, 2002). A functional polymorphism in intron 2 was associated with whole blood 5-HT levels in one study (Coutinho et al, 2004).
Recent studies suggest that other SLC6A4 polymorphisms may also be important in conferring risk for autism. Kim et al (2002) described two single-nucleotide polymorphisms (SNPs) (SNP10 [rs2020936], and SNP11 [rs2020937]) that were strongly associated with autism. A haplotype defined by T alleles at these two SNPs (SNP10-SNP11 TT) was most highly associated with autism (Cook EH, unpublished data). In a mutation screen of SLC6A4, Sutcliffe et al (2005) found rare functional SERT amino-acid changes that lead to increased 5-HT transport in subjects with autism (Prasad et al, 2005). Another amino-acid variant that leads to increased SERT function, Ile425Val, has been detected in subjects with obsessive–compulsive disorder (Ozaki et al, 2003; Delorme et al, 2005).
In our original study of first-degree relatives of patients with autism, hyperserotonemic subjects were found to have decreased 3H-LSD-marked receptor density (Cook et al, 1993). LSD binds to 5-HT receptors including 5-HT2A and 5-HT7, both of which are expressed in the platelet (Cook et al, 1994) (Cook EH, unpublished observation). Murphy et al (2006) recently described decreased cortical 5-HT2A binding in Asperger's syndrome, supporting the possible relevance of the platelet model. A functional amino-acid variant and a possibly functional promoter SNP have been described in the 5-HT2A gene (HTR2A) (Ozaki et al, 1997; Spurlock et al, 1998). One study found association at three adjacent polymorphisms in the first intron of HTR7 in autism (Prathikanti S and Cook EH, unpublished data).
Additional proteins may also be important in modulating the 5-HT system within the platelet. Variation in the integrin β3 gene (ITGB3) has been associated with platelet 5-HT in multiple populations, including probands with autism (Weiss et al, 2004, 2005b, 2006). Tryptophan hydroxylase 1 (TPH1), which catalyzes the rate-limiting step in 5-HT synthesis in the periphery, could also contain functional variation that would impact 5-HT synthesis.
Based on the platelet 5-HT indices in the two subpopulations of subjects with elevated platelet 5-HT, a series of hypotheses were generated. Polymorphisms and haplotypes in SLC6A4 were hypothesized to be associated with variation in Vmax, Km, and whole blood 5-HT levels. Polymorphisms in HTR7 and HTR2A were hypothesized to be associated with variation in LSD-binding and whole blood 5-HT levels. Finally, polymorphisms in ITGB3 and TPH1 were hypothesized to be associated with whole blood 5-HT levels. Our hypotheses were tested by genotyping the subjects previously studied for indices of the platelet serotonin system (Cook et al, 1993). Polymorphisms within each gene were selected based upon either reported association with autism or reported alteration in gene or protein function. In the case of TPH1, no known association with autism or functional variation has been reported, and two SNPs were chosen based upon position within a large haplotype block spanning the gene (Table 1).
METHODS
Subjects and Blood Collection
Subjects were selected as part of a previous study that measured indices of the platelet serotonin system (Cook et al, 1993). Two groups (normoserotonemic and hyperserotonemic) of 12 sex- and age-matched first-degree relatives of children with autism were studied (Table 2). Subjects were excluded if they had used psychotropic medication within 6 months of the study. Hyperserotonemia was defined as greater than 2 SDs above the mean as determined previously (Cook et al, 1988, 1993). Subjects were chosen based on the samples collected in February 1992. Blood was drawn and all platelet serotonin indices described below were conducted as part of the original published study (Cook et al, 1993). Although whole blood 5-HT levels are typically stable within individuals, some of the hyperserotonemic subjects showed instability and were not in the hyperserotonemic range when sampled at the time of the platelet assays, which may be related to the seasonal variation that has been described previously (Wirz-Justice et al, 1977; Hanna et al, 1998). The whole blood 5-HT level from the day of the platelet studies was used for analyses in the present study.
Whole Blood 5-HT and Platelet Count
Whole blood 5-HT was chosen as the most reliable measure of platelet 5-HT because greater than 99% of whole blood 5-HT is in the platelet fraction (Anderson et al, 1987). Direct measurement of platelet 5-HT by centrifugation adds laboratory error due to 5-HT release during processing or variable platelet yield. Whole blood 5-HT was measured by HPLC with fluorometric detection (Anderson et al, 1981). Intra-assay and interassay coefficients of variation were 1.7 and 6.2%, respectively. Platelet count was determined with a Thrombocounter C (Coulter Electronics, Hialeah, FL). Time of sampling may affect whole blood 5-HT levels (Wirz-Justice et al, 1977); therefore, all of the blood sampling was carried out between 0900 and 1000 hours. Similarly, an influence of seasonal variation on whole blood 5-HT levels has been described (Wirz-Justice et al, 1977), but samples for these studies were collected exclusively in late summer and early fall.
Platelet 5-HT Uptake and [3H]-LSD Binding
Platelet 5-HT uptake and [3H]-LSD binding were assayed in the laboratory of Dr Ramesh Arora at Hines V.A. Hospital as described previously (Cook et al, 1993). Six samples could not be assayed for platelet 5-HT uptake for logistical reasons because the assay had to be performed on the same day as the blood draw. One sample did not have sufficient tissue for [3H]LSD binding because of difficulty in blood drawing.
Platelet Count and Platelet 5-HT Uptake
Platelets were counted electronically by means of a Coulter thrombocounter in triplicate. 5-HT uptake was studied as described earlier (Arora and Meltzer, 1981). Briefly, platelet-rich plasma (0.3 ml) was preincubated with Kreb's phosphate buffer (0.6 ml, pH 7.4) minus CaCl2 for 10 min at 37°C. Next 0.1 ml of [14C]5-HT was added to yield final concentrations of 5-HT of 0.3, 0.5, 0.75, and 1 μM. One set of tubes was immediately immersed in an ice bath while the other set was incubated for an additional 4 min. Radioactivity in the platelet pellet was counted as described earlier (Arora and Meltzer, 1981).
[3H]-LSD Binding
A 0.2 ml aliquot containing 100–150 μg of membrane protein was incubated with [3H]-LSD (specific activity 79.9 Ci/mmol) in Tris HCl+NaCl+KCl buffer (pH 7.4) for 4 h at 37°C in the presence and absence of spiperone (300 nM). After incubation, the reaction was terminated by the addition of Tris-HCl (pH 7.7, 50 mM) containing 0.1% bovine serum albumin and rapidly filtered through GF/F filters. The filters were washed and the radioactivity was counted as described previously (Cook et al, 1993).
The specific binding of [3H]-LSD was defined as the difference in binding in the presence and absence of spiperone. Five to six concentrations of [3H]LSD (0.2–4 nM) were used in the incubation mixture to determine the dissociation constant (Kd) and the density of [3H]-LSD biding sites (Bmax) by Scatchard analysis.
DNA Isolation
DNA was isolated from 300 μl of frozen whole blood samples using the Puregene® DNA Purification method (Gentra Systems, Minneapolis, MN, http://www.gentra.com).
Genotyping
Single-Nucleotide Polymorphisms
Nine SNP markers were genotyped using the TaqMan® assay (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com). Six SNP markers were genotyped using the Assays-on-Demand™ with standard conditions (HTR2A rs6311/C_8695278, HTR7 rs2185706/ C_11756741, HTR7 C_416286, HTR7 C_8418463, TPH1 rs1799913/C_2645661, and TPH1 rs623580/C_2645676; whereas four SNP markers were genotyped using TaqMan® Assays-by-Design (HTR2A rs6314, SLC6A4-Ile425Val, SLC6A4-Gly56Ala, SLC6A4 SNP10/rs2020936). A control sample known to contain the SLC6A4 Ile425Val variant (Glatt et al, 2001) from the DNA Polymorphism Discovery Resource (DPDR, http://locus.umdnj.edu/nigms/pdr.html) was used to confirm assay performance. Samples known to contain the SLC6A4-Gly56Ala variant were also genotyped to confirm assay performance (Sutcliffe et al, 2005). One subject was not genotyped for the two TPH1 SNPs due to scarcity of DNA. Primers and probes for Assays-By-Design are shown in Table 3.
One SNP marker (SLC6A4 SNP11/rs2020937) was genotyped by Fluorescence Polarization-Template Directed dye-terminator Incorporation assay (FP-TDI) (Chen et al, 1999) as described previously (Kim et al, 2002).
The ITGB3 Leu33Pro SNP (rs5918) was genotyped with a restriction fragment length polymorphism assay using MspI (New England Biolabs, http://www.neb.com) as described previously (Weiss et al, 2006).
Length Polymorphisms
The SLC6A4 HTTLPR and intron 2 VNTR polymorphisms were amplified and detected as described previously (Kim et al, 2002).
Statistical Analysis
Haplotypes at the four common SLC6A4 polymorphisms were predicted using web-based PHASE version 2.0. (Stephens et al, 2001; Stephens and Donnelly, 2003). Five haplotypes were predicted (Table 4). These were condensed to three groups of diplotypes using a priori predictions from analyses in other populations. Group 1 was homozygous for haplotypes containing the T allele at both SLC6A4 SNP10 and SNP11. The SNP10-SNP11 TT haplotype was most highly associated with autism in a dense association mapping study (Cook EH, unpublished analysis of data from Kim et al (2002)), and we reasoned that it may be separately associated with altered transporter function and elevated whole blood 5-HT. Group 2 was homozygous for haplotypes containing the long allele of the HTTLPR polymorphism previously associated with elevated platelet 5-HT uptake (Greenberg et al, 1999; Anderson et al, 2002). Group 3 had other combinations of haplotypes. These three groups were compared by ANOVA for platelet 5-HT maximum uptake rate (Vmax), platelet 5-HT affinity (Km), and whole blood 5-HT levels. When significant association was detected, post hoc analysis by ANOVA compared three groups: group 1 consisted of subjects homozygous for haplotypes containing the T allele at both SNP10 and SNP11, group 2 consisted of subjects heterozygous for haplotypes containing the T allele at these two SNPs, and group 3 consisted of subjects without haplotypes containing the T allele at these two SNPs. Follow-up t-test was used to compare subjects homozygous for haplotypes containing the T allele at both SNP10 and SNP11 and all other subjects. When significant association was observed, follow-up analysis was performed excluding the four non-Caucasian subjects to remove potential population stratification bias. Post hoc analysis by ANOVA was also conducted to compare the three genotype groups for each individual SLC6A4 marker.
Genotypes at each HTR2A and HTR7 polymorphism were assessed for association with [3H]-LSD binding and whole blood 5-HT using ANOVA and independent sample t-test. The HTR2A His452Tyr variant was only observed in three subjects, and haplotype analysis was not performed for the two HTR2A SNPs. The three HTR7 SNPs (C_416286, C_8418436, and rs2185706) were found to be in perfect linkage disequilibrium (LD). Results reported for rs2185706, therefore, also apply to the other two HTR7 SNPs. Haplotypes at the two TPH1 SNPs were constructed by using web-based PHASE version 2.0 (Table 5) (Stephens et al, 2001; Stephens and Donnelly, 2003). Allele-wise haplotype analysis by ANOVA was used to compare whole blood 5-HT across the three observed haplotypes. Genotype at both TPH1 SNPs was assessed for association with whole blood 5-HT using independent sample t-test. When significant association between TPH1 haplotypes and whole blood 5-HT was detected, follow-up analyses were performed to examine potential sources of bias. To correct for possible effects of platelet count, three subjects with outlier platelet count values (below 150 000 or above 450 000 platelets per microliter) were eliminated from the analysis of TPH1 haplotype effects and whole blood 5-HT per platelet was used as the dependent measure. To correct for possible effects of ethnicity, the four non-Caucasian subjects were eliminated from the analysis. Allele-wise haplotype analysis by ANOVA was used to compare whole blood 5-HT across the three observed haplotypes in the remaining 17 subjects.
Association at individual SNPs was assessed by ANOVA, or by t-test when only two genotype groups were present (rs6314) or when there were fewer than five subjects with a particular genotype (rs2185706, rs623580, and rs1799913). In the case of a homozygous genotype occurring fewer than five times, subjects were lumped with heterozygotes for statistical analysis. Bonferroni correction was not applied to these exploratory data. The Kolmogorov–Smirnov test was used to verify that each dependent variable did not deviate from normality.
RESULTS
Platelet 5-HT Uptake Vmax and Km
ANOVA comparing SNP10-SNP11 TT/TT homozygotes, HTTLPR L/L homozygotes, and subjects with other haplotype combinations, revealed a significant effect on Km (F=7.75, p=0.005, n=18) and Vmax (F=3.82, p=0.046, n=18) across the three groups (see Figure 1). Follow-up analysis excluding the four non-Caucasian subjects confirmed a significant effect on Km (F=5.770, p=0.018, n=14) but did not show a significant effect on Vmax (F=2.539, p=0.120, n=14). Of note, all of the subjects in the SNP10-SNP11 TT/TT homozygote group had HTTLPR S/S or S/L genotypes. Post hoc ANOVA comparing SNP10-SNP11 TT/TT homozygotes, SNP10-SNP11 TT heterozygotes, and subjects with other haplotypes revealed a significant effect on Km (F=6.29, p=0.01) but not on Vmax (F=2.726, p=0.098). Follow-up analysis excluding the four non-Caucasian subjects confirmed a significant effect on Km (F=4.315, p=0.039) but again not on Vmax (F=2.217, p=0.152). T-test demonstrated that SNP10-SNP11 TT/TT homozygotes differed significantly from other subjects for Km (p=0.002) and Vmax (p=0.029). Follow-up analysis excluding the four non-Caucasian subjects confirmed a significant effect on Km (p=0.009) but revealed only a supportive trend for Vmax (p=0.053). ANOVAs for genotypes of each individual marker, including the HTTLPR, revealed no significant effects.

Altered Km and Vmax by SLC6A4 diplotype group. Mean values with SEM of (a) [14C]-5-HT binding affinity Km and (b) [14C]-5-HT uptake Vmax are shown for each of three SLC6A4 diplotype groups. Subjects in the TT/TT group had two haplotypes (ie a diplotype) containing the SNP10–SNP11 T–T haplotype. All of these subjects had HTTLPR S/S or S/L genotypes. Subjects in the L/L group had two haplotypes containing the HTTLPR L allele. Subjects in the ‘other’ group had other combinations of haplotypes and all had HTTLPR S/S or S/L genotypes.
[3H]LSD Binding
Genotypes at the HTR7 LD block and the two HTR2A polymorphisms were analyzed by ANOVA (rs6311) or independent sample t-test (rs6314 and rs2185706) for association with [3H]-LSD binding (Bmax). Although neither HTR2A polymorphism was found to be significantly associated with [3H]LSD binding (rs6311 F-value=1.431, p=0.263 and rs6314 t-value=0.736, p=0.470, n=23) a trend was observed for the HTR7 SNP (rs2185706 t-value=1.944, p=0.065).
Whole Blood 5-HT
Analysis with PHASE generated three haplotypes between the two TPH1 SNPs, indicating that these two SNPs are in complete, but not perfect, LD in this sample. Data from the HapMap project show a region of high LD in this region, including the entire TPH1 gene (D′=0.92–1.0; r2=0.10–1.0; see Table 1) (The International HapMap Consortium, 2003). Allele-wise haplotype analysis by ANOVA revealed a significant effect on whole blood 5-HT (F=3.31, p=0.046, n=23, see Figure 2). This effect remained significant after correction for platelet count and removal of non-Caucasian subjects and those with outlier platelet values (F=3.991, p=0.029, n=17). Follow-up ANOVA applied to subjects with 0, 1, or 2 copies of the G–A haplotype revealed a significant effect (F=3.75, p=0.041). This effect also remained significant after correction for platelet count and removal of non-Caucasian subjects and those with outlier platelet values (F=6.267, p=0.011). Genotypes at the two individual TPH1 polymorphisms were analyzed by independent sample t-test (rs623580 and rs1799913) for association with whole blood 5-HT, and a trend was observed for association with whole blood 5-HT at each SNP (rs623580, p=0.054; rs1799913, p=0.059). No significant effects of SLC6A4 polymorphisms or haplotypes, or HTR2A, HTR7, or ITGB3 genotypes on whole blood 5-HT were detected by ANOVA.

Altered whole blood 5-HT by TPH1 diplotype group. Mean values with SEM of whole blood 5-HT are shown for subjects with three different TPH1 diplotypes, containing either none, one, or two copies of the rs1799913–rs623580 haplotype.
Uncommon Functional SERT Variants
The Gly56Ala variant was detected in two normoserotonemic subjects and no hyperserotonemic subjects. No relationship was seen between this variant and whole blood 5-HT, Km, or Vmax. The Ile425Val variant was not present in our sample.
DISCUSSION
Hyperserotonemia occurs in approximately one-third of patients with autism (Cook and Leventhal, 1996) and is also familial, occurring in some first-degree relatives (Abramson et al, 1989; Cook et al, 1990). The association tests presented here are a continuing effort to understand these abnormalities in the 5-HT system. Unlike studies of candidate genes in the disorder itself, polymorphisms in candidate genes were studied in relationship to platelet 5-HT measures.
Significant differences were observed between different haplotype groups at SLC6A4 for transporter Km and Vmax. Appropriate statistical correction for multiple testing is difficult to assess when analyzing multiple polymorphisms in LD with one another, but only the Km association appears robust to correction. Post hoc analysis revealed that the difference in Km was largely contained in the contrast between subjects homozygous for the SNP10-SNP11 TT haplotype and other subjects (p=0.002). This is especially remarkable because these subjects all had HTTLPR S/S or S/L genotypes, which have been previously associated with lower 5-HT transport Vmax in comparison with the L/L genotype (Greenberg et al, 1999; Anderson et al, 2002). This association suggests that the TT haplotype may be in LD with a functional variant or variants elsewhere in the gene or may itself have some unknown functional significance. The functional variant(s) could alter gene expression, protein trafficking, or protein regulation (Lesch et al, 1996; Prasad et al, 2005), and may demonstrate its platelet phenotype indirectly as a regulatory response to altered SERT structure or function (Jayanthi et al, 2005; Zhu et al, 2005; Carneiro and Blakely, 2006). The two most commonly observed amino-acid changes, Gly56Ala and Ile425Val, do not account for the association in this sample. Interestingly, although the SNP10-SNP11 TT haplotype is significantly associated with transporter function, it does not show a significant association with whole blood 5-HT itself.
The seeming contradiction of altered transporter function without altered platelet 5-HT levels points to a heterogeneous etiology of hyperserotonemia in autism, as predicted from the original study in which transporter Vmax and LSD binding were elevated and decreased, respectively, in separate subgroups (Cook et al, 1993; Cook and Leventhal, 1996). Seasonal variation of platelet 5-HT, which has previously been described in association with the SLC6A4 HTTLPR polymorphism (Hanna et al, 1998), could also add further complexity to the relationship between 5-HT levels and 5-HT transport function. It is possible that multiple polymorphisms at SLC6A4 have varying effects on function that may interact with one another in ways that we cannot yet predict. These variants may include both common polymorphisms of small effect, such as HTTLPR or the intron 2 VNTR, as well as rare variants of larger effect, such as the Ile425Val variant. Our inability to detect a platelet phenotype in the two normoserotonemic subjects with the SERT Gly56Ala variant is not altogether surprising because this variant showed only one-third elevated 5-HT transport compared to the wild type in a cellular model (Prasad et al, 2005). A preponderance of other uncommon variation in the gene region suggests that other variants may also affect transcription or trafficking of SLC6A4 (Sutcliffe et al, 2005). The genetic component of familial hyperserotonemia in autism also likely includes the effects of variants in other genes, whether tested here or still unknown.
Another independent contributor to familial hyperserotonemia in autism is altered 5-HT receptor binding. Two HTR2A polymorphisms showed no association with [3H]LSD binding. Analysis of possible parent-of-origin effects was not possible for this variably imprinted gene (Kato et al, 1996, 1998; Bunzel et al, 1998). Polymorphisms in HTR7 are also a potential source of altered [3H]-LSD binding. A recent study found a nominal association between a set of three polymorphisms in strong LD with one another and autism (Prathikanti S and Cook EH, unpublished data). These polymorphisms were observed to be in perfect LD in our population and showed a trend toward association with [3H]LSD binding.
Platelet hyperserotonemia could also be explained by variation in other proteins known to be important within the 5-HT system. Previous studies have found association between ITGB3 and whole blood 5-HT levels in males (Weiss et al, 2004, 2005a, 2005b), but our study is too small to support a sex-specific analysis. Additionally, allelic heterogeneity with regard to 5-HT levels is evident at ITGB3. The amino-acid polymorphism that we genotyped in this study showed evidence for association with autism in previous studies, but did not show the strongest evidence for association with serotonin level in these studies (Weiss et al, 2005b, 2006). A haplotype of two TPH1 polymorphisms was associated with whole blood 5-HT by allele-wise ANOVA (p=0.046). These nominally significant findings would not survive statistical correction for multiple testing. If replicated, this association would suggest that TPH1 variation may have important effects on platelet 5-HT synthesis in the periphery (Walther et al, 2003). These two SNPs are unlikely to be functional themselves, but high LD in this region suggests that they would be likely to be in LD with any common functional variation in this region (The International HapMap Consortium, 2003). Although it is not responsible for the majority of central 5-HT synthesis in the mature animal, TPH1 remains a candidate for developmental disorders because of its role in 5-HT synthesis late in brain development (Nakamura et al, 2006).
The primary limitation of this study is sample size. We have limited power to detect association between genotype and indices of the platelet 5-HT system. A second limitation is the absence of family genotype data or genomic control in a quantitative genotype comparison study. Methodology has not yet been developed for genomic control when considering association with quantitative traits. Analysis including only Caucasian subjects supported the findings in the overall group of subjects.
These results build on previous research that found familial hyperserotonemia in autism to be associated with either increased 5-HT uptake or decreased receptor binding. Variation in SLC6A4 was associated with 5-HT binding and uptake but did not have a significant effect on whole blood 5-HT. Genotypes at HTR7 polymorphisms previously associated with autism need further study in relation to [3H]-LSD binding. Haplotypes at two polymorphisms in TPH1 may be associated with whole blood 5-HT levels. The platelet hyperserotonemia story remains complex and will require additional study in a larger population. Complete characterization of the variation around the SLC6A4 gene in a sample of patients or first-degree relatives with hyperserotonemia may be necessary to clarify the role of multiple potentially functional variants. Our data point to variation in other mechanisms that may play a larger role in hyperserotonemia than 5-HT uptake itself. Denser genotyping of polymorphisms and resequencing studies of TPH1, ITGB3, and HTR7 may also be necessary to clarify their roles and identify functional variants that may be relevant to hyperserotonemia. Finally, a larger sample will enable analysis of possible allelic or gene–gene interaction within the peripheral serotonin system.
References
Abramson RK, Wright HH, Carpenter R, Brennan W, Lumpuy O, Cole E et al (1989). Elevated blood serotonin in autistic probands and their first-degree relatives. J Autism Dev Disord 19: 397–407.
Anderson GM, Feibel FC, Cohen DJ (1987). Determination of serotonin in whole blood, platelet-rich plasma, platelet-poor plasma and plasma ultrafiltrate. Life Sci 40: 1063–1070.
Anderson GM, Gutknecht L, Cohen DJ, Brailly-Tabard S, Cohen JH, Ferrari P et al (2002). Serotonin transporter promoter variants in autism: functional effects and relationship to platelet hyperserotonemia. Mol Psychiatry 7: 831–836.
Anderson GM, Young JG, Cohen DJ, Schlicht KR (1981). Liquid-chromatographic determination of serotonin and tryptophan in whole blood and plasma. Clin Chem 27: 775–776.
Arora R, Meltzer H (1981). A modified assay for determining serotonin uptake in human platelets. Clin Chem Acta 112: 225–233.
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E et al (1995). Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 25: 63–78.
Bunzel R, Bluemcke I, Cichon S, Normann S, Schramm J, Propping P et al (1998). Polymorphic imprinting of the serotonin-2A (5-HT2A) receptor gene in human adult brain. Mol Brain Res 59: 90–92.
Carneiro AM, Blakely RD (2006). Serotonin, protein kinase C and HIC-5 associated redistribution of the platelet serotonin transporter. J Biol Chem 281: 24769–24780.
Chen X, Levine L, Kwok P (1999). Fluorescence polarization in homogeneous nucleic acid analysis. Genome Res 9: 492–498.
Cook E, Arora R, Anderson G, Berry-Kravis E, Yan S-Y, Yeoh H et al (1993). Platelet serotonin studies in hyperserotonemic relatives of children with autistic disorder. Life Sci 52: 2005–2015.
Cook E, Leventhal B (1996). The serotonin system in autism. Curr Opin Pediatr 8: 348–354.
Cook EH, Fletcher KE, Wainwright M, Marks N, Yan S-Y, Leventhal BL (1994). Primary structure of the human platelet serotonin 5-HT2A receptor: Identity with frontal cortex serotonin 5-HT2A receptor. Neurochem 63: 465–469.
Cook EH, Leventhal BL, Freedman DX (1988). Serotonin and measured intelligence. J Autism Dev Disord 18: 553–559.
Cook EH, Leventhal BL, Heller W, Metz J, Wainwright M, Freedman DX (1990). Autistic children and their first-degree relatives: Relationships between serotonin and norepinephrine levels and intelligence. J Neuropsychiatry Clin Neurosci 2: 268–274.
Coutinho AM, Oliveira G, Morgadinho T, Fesel C, Macedo TR, Bento C et al (2004). Variants of the serotonin transporter gene (SLC6A4) significantly contribute to hyperserotonemia in autism. Mol Psychiatry 9: 264–271.
Delorme R, Betancur C, Wagner M, Krebs MO, Gorwood P, Pearl P et al (2005). Support for the association between the rare functional variant I425V of the serotonin transporter gene and susceptibility to obsessive compulsive disorder. Mol Psychiatry 10: 1059–1061.
Devlin B, Cook EH, Coon H, Dawson G, Grigorenko EL, McMahon W et al (2005). Autism and the serotonin transporter: the long and short of it. Mol Psychiatry 10: 1110–1116.
Folstein S, Rutter M (1977). Genetic influences and infantile autism. Nature 265: 726–728.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B et al (2002). The structure of haplotype blocks in the human genome. Science 296: 2225–2229.
Glatt C, DeYoung J, Delgado S, Service S, Giacomini K, Edwards R et al (2001). Screening a large reference sample to identify very low frequency sequence variants: comparisons between two genes. Nat Genetics 27: 435–438.
Greenberg B, Tolliver T, Huang S, Li Q, Bengel D, Murphy D (1999). Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet (Neuropsychiatric Genet) 88: 83–87.
Hanna GL, Himle JA, Curtis GC, Koram DQ, Weele JVV, Leventhal BL et al (1998). Serotonin transporter and seasonal variation in blood serotonin in families with obsessive-compulsive disorder. Neuropsychopharmacology 18: 102–111.
Jayanthi LD, Samuvel DJ, Blakely RD, Ramamoorthy S (2005). Evidence for biphasic effects of protein kinase C on serotonin transporter function, endocytosis, and phosphorylation. Mol Pharmacol 67: 2077–2087.
Kato MV, Ikawa Y, Hayashizaki Y, Shibata H (1998). Paternal imprinting of mouse serotonin receptor 2A gene Htr2 in embryonic eye: a conserved imprinting regulation on the RB/Rb locus. Genomics 47: 146–148.
Kato MV, Shimizu T, Nagayoshi M, Kaneko A, Sasaki MS, Ikawa Y (1996). Genomic imprinting of the human serotonin-receptor (HTR2) gene involved in development of retinoblastoma. Am J Hum Genet 59: 1084–1090.
Kim S-J, Cox N, Courchesne R, Lord C, Corsello C, Akshoomoff N et al (2002). Transmission disequilibrium mapping in the serotonin transporter gene (SLC6A4) region in autistic disorder. Mol Psychiatry 7: 278–288.
Lesch K-P, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S et al (1996). Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274: 1527–1531.
Murphy DG, Daly E, Schmitz N, Toal F, Murphy K, Curran S et al (2006). Cortical serotonin 5-HT2A receptor binding and social communication in adults with Asperger's syndrome: an in vivo SPECT study. Am J Psychiatry 163: 934–936.
Nakamura K, Sugawara Y, Sawabe K, Ohashi A, Tsurui H, Xiu Y et al (2006). Late developmental stage-specific role of tryptophan hydroxylase 1 in brain serotonin levels. J Neurosci 26: 530–534.
Ozaki N, Goldman D, Kaye W, Plotnikov K, Greenberg B, Rudnick G et al (2003). A missense mutation in the serotonin transporter is associated with a complex neuropsychiatric phenotype. Mol Psychiatry 8: 933–936.
Ozaki N, Manji H, Lubierman V, Lu SJ, Lappalainen J, Rosenthal NE et al (1997). A naturally occurring amino acid substitution of the human serotonin 5- HT2A receptor influences amplitude and timing of intracellular calcium mobilization. J Neurochem 68: 2186–2193.
Piven J, Tsai G, Nehme E, Coyle JT, Folstein SE (1991). Platelet serotonin, a possible marker for familial autism. J Autism Dev Disord 21: 51–59.
Prasad HC, Zhu CB, McCauley JL, Samuvel DJ, Ramamoorthy S, Shelton RC et al (2005). Human serotonin transporter variants display altered sensitivity to protein kinase G and p38 mitogen-activated protein kinase. Proc Natl Acad Sci USA 102: 11545–11550.
Schain RJ, Freedman DX (1961). Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J Pediatrics 58: 315–320.
Spurlock G, Heils A, Holmans P, Williams J, D’Souza UM, Cardno A et al (1998). A family based association study of T102C polymorphism in 5HT2A and schizophrenia plus identification of new polymorphisms in the promoter. Mol Psychiatry 3: 42–49.
Stephens M, Donnelly P (2003). A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73: 1162–1169.
Stephens M, Smith NJ, Donnelly P (2001). A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68: 978–989.
Stone JL, Merriman B, Cantor RM, Yonan AL, Gilliam TC, Geschwind DH, Nelson SF (2004). Evidence for sex-specific risk alleles in autism spectrum disorder. Am J Hum Genet 75: 1117–1123.
Sutcliffe JS, Delahanty RJ, Prasad HC, McCauley JL, Han Q, Jiang L et al (2005). Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid-compulsive behaviors. Am J Hum Genet 77: 265–279.
The International HapMap Consortium (2003). The International HapMap Project. Nature 426: 789–796.
Walther DJ, Peter JU, Bashammakh S, Hortnagl H, Voits M, Fink H et al (2003). Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299: 76.
Weiss LA, Abney M, Cook Jr EH, Ober C (2005a). Sex-specific genetic architecture of whole blood serotonin levels. Am J Hum Genet 76: 33–41.
Weiss LA, Abney M, Parry R, Scanu AM, Cook Jr EH, Ober C (2005b). Variation in ITGB3 has sex-specific associations with plasma lipoprotein(a) and whole blood serotonin levels in a population-based sample. Hum Genet 117: 81–87.
Weiss LA, Kosova G, Delahanty RJ, Jiang L, Cook EH, Ober C et al (2006). Variation in ITGB3 is associated with whole-blood serotonin level and autism susceptibility. Eur J Hum Genet 14: 923–931.
Weiss LA, Veenstra-VanderWeele J, Abney M, Newman DL, Kim S-J, Dytch H et al (2004). Genome-wide association study identifies ITGB3 as a QTL for whole blood serotonin. Eur J Hum Genet 12: 949–954.
Wirz-Justice A, Lichtsteiner M, Feer H (1977). Diurnal and seasonal variations in human platelet serotonin in man. J Neural Transm 41: 7–15.
Yonan AL, Alarcon M, Cheng R, Magnusson PK, Spence SJ, Palmer AA et al (2003). A genomewide screen of 345 families for autism-susceptibility loci. Am J Hum Genet 73: 886–897.
Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD (2005). p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem 280: 15649–15658.
Acknowledgements
Kathy Hennessy, Diane Dickel, and Laura Martinolich provided expert technical assistance. Charles Glatt generously provided information about the DPDR subject heterozygous for the SLC6A4 amino-acid variant. We are especially grateful to the subjects who participated in the study. This work was supported, in part, by NIH K02 MH01389 (EHC), NIH K20 MH01065 (GLH), NIH NS049261 (JSS), NIH T35 DK62719 (SC), the Jean Young and Walden W Shaw Foundation (BLL), the Harris Foundation (BLL), the Brain Research Foundation (EHC), University of Chicago, and the American Psychiatric Institute for Research and Education Janssen Scholars in Research on Severe Mental Illness program (JV-VW).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cross, S., Kim, SJ., Weiss, L. et al. Molecular Genetics of the Platelet Serotonin System in First-Degree Relatives of Patients with Autism. Neuropsychopharmacol 33, 353–360 (2008). https://doi.org/10.1038/sj.npp.1301406
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301406
Keywords
This article is cited by
-
Investigation of Genetic Polymorphism in Autism Spectrum Disorder: a Pathogenesis of the Neurodevelopmental Disorder
Advances in Neurodevelopmental Disorders (2022)
-
Intestinal Predictors of Whole Blood Serotonin Levels in Children With or Without Autism
Journal of Autism and Developmental Disorders (2022)
-
A perspective on molecular signalling dysfunction, its clinical relevance and therapeutics in autism spectrum disorder
Experimental Brain Research (2022)
-
Serotonin transporter availability in adults with autism—a positron emission tomography study
Molecular Psychiatry (2021)
-
Platelet studies in autism spectrum disorder patients and first-degree relatives
Molecular Autism (2015)
