
Abstract
Fueled by anatomical, electrophysiological, and pharmacological analyses of endogenous brain reward systems, norepinephrine (NE) was identified as a key mediator of both natural and drug-induced reward in the late 1960s and early 1970s. However, reward experiments from the mid-1970s that could distinguish between the noradrenergic and dopaminergic systems resulted in the prevailing view that dopamine (DA) was the primary ‘reward transmitter’ (a belief holding some sway still today), thereby pushing NE into the background. Most damaging to the NE hypothesis of reward were studies demonstrating that NE receptor antagonists and NE reuptake inhibitors failed to impact drug self-administration. In recent years new tools, such as genetically engineered mice, and new experimental paradigms, such as reinstatement of drug seeking following withdrawal, have propelled NE back into the awareness of addiction researchers. Of particular interest is disulfiram, an inhibitor of the NE biosynthetic enzyme dopamine β-hydroxylase, which has demonstrated promising efficacy in the treatment of cocaine dependence in preliminary clinical trials. The purpose of this review is to synthesize the new data linking NE to critical aspects of DA signaling and drug addiction, with a focus on psychostimulants (eg, cocaine), opiates (eg, morphine), and alcohol.
Similar content being viewed by others

INTRODUCTION
Norepinephrine (NE) is one of the most abundant neurotransmitters in the brain, where it plays an important role in selective attention, general arousal, and stress reactions in challenging environments (Foote et al, 1983; Levine et al, 1990; Berridge and Waterhouse, 2003; Aston-Jones and Cohen, 2005). NE has also been implicated in diverse central processes and diseases, including learning and memory, neuronal excitability, pain, and affective disorders (Ressler and Nemeroff, 1999; Gibbs and Summers, 2002; Jasmin et al, 2002; Weinshenker et al, 2001; Murchison et al, 2004).
The brain noradrenergic system is comprised of two main ascending projections: the dorsal noradrenergic bundle (DNB), which originates in the A6 locus coeruleus (LC) and projects to the hippocampus, cerebellum, and forebrain, and the ventral noradrenergic bundle (VNB), which arises in a number of nuclei of the pons and medulla, such as the A1 and A2 cell groups, and innervates the hypothalamus, midbrain, and extended amygdala (reviewed by Moore and Bloom, 1979). These neuroanatomical substrates underlie NE's ability to impinge upon brain systems that control multiple aspects of drug addiction, including sensitization, reward, and relapse.
This review is comprised of four main parts. In the first, we will summarize the early studies that identified NE as an important mediator of drug reward, as well as the subsequent experiments implicating dopamine (DA) that led to the downfall of the noradrenergic theory of reward. Second, we will review the recent literature placing NE at the forefront again as a critical mediator of drug reward and the addiction process, with a focus on psychostimulants, opiates, and ethanol. Third, we will catalog the interplay between the noradrenergic and dopaminergic systems underlying at least some of the effects of NE on drug reward. Finally, we will review clinical studies assessing the effects of noradrenergic gene polymorphisms on drug responses and the efficacy of compounds that modulate NE signaling for the treatment of drug addiction.
PART I: THE RISE AND FALL OF THE NORADRENERGIC THEORY OF DRUG REWARD
NE was implicated as a key mediator of drug reward for three primary reasons: noradrenergic pathways support intracranial self-stimulation (ICSS) and modulate drug-induced changes in ICSS threshold; the biochemical activity of psychostimulant drugs includes blockade of NE reuptake and enhancement of NE release; and compounds that interfere with NE synthesis or signaling influence drug self-administration (SA).
NE and Intracranial Self-Stimulation
ICSS, which refers to the ability of animals to operantly administer electrical stimulation to the brain, was first described by Olds and Milner (1954). The fact that electrodes placed in some regions of the brain (but not others) could support ICSS implied that anatomically specialized reward systems existed in the brain, and many subsequent studies were designed to dissect the neurochemical basis of these reward pathways (Wise, 1978). NE was first hypothesized as a key reward neurotransmitter in the early 1960s (Poschel and Ninteman, 1963; Stein, 1964)—a hypothesis that was supported when self-stimulation sites were found in the LC and along the DNB projection path (Dresse, 1966; Crow et al, 1972; Ritter and Stein, 1973). Positive self-stimulation sites were also found along the VNB (Ritter and Stein, 1974). Furthermore, NE is released during medial forebrain bundle (MFB) ICSS, and central administration of NE facilitates MFB ICSS (Stein and Wise, 1969; Wise and Stein, 1969; Wise et al, 1973). Finally, drugs that deplete NE stores, interfere with NE synthesis, or ablate NE neurons disrupt ICSS (reviewed by Fibiger and Phillips, 1974; Wise, 1978).
The idea that drugs of abuse act via the endogenous reward systems in the brain first arose when psychostimulants (eg, amphetamine, cocaine) were found to alter ICSS thresholds (Stein, 1964; Crow, 1970; Wise, 1978). One interpretation of these results was that NE mediates the effect of psychostimulants on ICSS, because these drugs cause NE release, block NE reuptake, or both.
Yet despite evidence supporting the role of NE in ICSS, a number of subsequent studies cast doubt on its importance. Some groups were unable to reproduce the original finding that the LC could support self-stimulation (Amaral and Routtenberg, 1975; Simon et al, 1975). Furthermore, 6-hydroxydopmaine (6-OHDA) or electrolytic lesions of the DNB failed to attenuate LC self-stimulation, and LC lesions failed to disrupt self-stimulation of sites along the DNB. ICSS was also unaffected by administration of antagonists at low doses that are selective for adrenergic receptors (reviewed by Wise, 1978). Taken together, these findings suggest that, whereas NE might contribute to ICSS and reward, it is by no means a critical component of this system.
Contemporaneously with the above studies, a number of groups were investigating the possibility that DA, not NE, was the critical neurotransmitter mediator of ICSS and endogenous reward. These studies have been extensively reviewed elsewhere (eg, Wise, 1978), but to summarize, DA passed many of the criteria that NE failed. For example, self-stimulation sites were also found at DA cell bodies and projection fields, and nearly all the pharmacological agents used to implicate NE (eg, reserpine, 6-OHDA, amphetamine, cocaine, and α-methyl-p-tyrosine (AMPT)) also affect dopaminergic systems. Most importantly, selective destruction of DA neurons or blockade of DA receptors profoundly attenuate ICSS. By the late 1970s DA, rather than NE, was generally accepted as the brain's primary ‘reward neurotransmitter’ in the context of ICSS.
NE and Psychostimulant SA
Rodents and nonhuman primates will perform operant tasks (eg, lever press, nose poke) for intravenous and intracranial injections of drugs that are abused by humans, including psychostimulants (eg, cocaine, amphetamine), and opiates (eg, morphine, heroin). Experiments using alcohol typically involve operant or voluntary oral ethanol ingestion. Collectively, these ‘SA paradigms are generally considered the gold standard for assessing the reinforcing properties of a drug. There are four distinct phases of SA: acquisition (when the animal learns the operant behavior), maintenance (when drug intake patterns are stable), extinction (when the operant behavior is extinguished by substitution of an inactive solution for the drug), and reinstatement (when the operant behavior is restored by contextual cues, stress, or drug priming). Until recently, the maintenance phase was usually the only one used to assess and interpret addiction pathways, and this phase is often synonymous with SA itself.
Relatively early on, catecholamines were found to be critical for psychostimulant SA, as catecholamine synthesis inhibitors or nonselective catecholamine receptor antagonists produced effects similar to reward reduction and termination (reviewed by Wise, 1978). When treatments that could distinguish between the contributions of noradrenergic and dopaminergic systems were tested, it became clear that DA, not NE, was the primary mediator of psychostimulant SA. For example, DA receptor antagonists increase the response rate for amphetamine or cocaine, whereas NE receptor antagonists have little or no effect (Yokel and Wise, 1975, 1976; Woolverton, 1987). Two studies (Goldberg and Gonzalez, 1976; Harris et al, 1996) demonstrated a decrease in cocaine SA following treatment with propanolol, a β-antagonist, but there was a concomitant decrease in responding for food in the latter study, suggesting a nonspecific effect on task performance. Although 6-OHDA lesions of DA produce a long-lasting reduction in SA of cocaine, lesions of both the dorsal and VNBs fail to alter responding (Roberts et al, 1977). Furthermore, while selective DA reuptake inhibitors themselves are readily self-administered and alter psychostimulant SA, selective NET inhibitors possess neither property (Woolverton, 1987; Howell and Byrd, 1991; Skjoldager et al, 1993; Tella, 1995; Wee et al, 2006). The only noradrenergic drug that appears to have reinforcing properties is clonidine, an α2-adrenergic receptor (α2AR) agonist that is self-administered by both rats and nonhuman primates (Shearman et al, 1981; Woolverton et al, 1982). The α2ARs are located on noradrenergic neurons, where they function as inhibitory autoreceptors, as well as on the dendrites and terminals of NE target cells—the latter population of α2ARs appears to mediate the rewarding effects of clonidine (Cervo et al, 1993). The mechanism by which clonidine produces its reinforcing effects is not clear. However, it is interesting to note that α2ARs, especially the α2cAR subtype, are highly expressed in striatum and can be activated by DA. Given the relatively sparse innervation of the striatum by noradrenergic neurons (especially the dorsal striatum), the suggestion is that DA is the primary endogenous ligand for α2ARs in the striatum (Zhang et al, 1999). Perhaps the rewarding effects of clonidine are mediated by direct activation of striatal α2ARs, which likely converge on some of the striatal signaling pathways activated by DA—a hypothesis that could be tested directly by assessing SA of clonidine directly into the striatum.
As a whole, these studies solidified the DA hypothesis of drug reward whereas refuting an important role for NE, and NE naturally faded from the consciousness of most psychostimulant addiction researchers until the late 1990s. However, there are some crucial points that must be emphasized here. First, all the SA studies we have mentioned examined one phase of SA only: maintenance of an established behavior. The influences of NE on acquisition, extinction, and reinstatement of drug-seeking behavior, which model other critical aspects of drug addiction, were not tested. There are more recent studies (to be discussed later) that have strongly implicated NE as a critical mediator of reinstatement of an extinguished SA. The second point is that SA is only one way of measuring drug reward. As will be seen in subsequent sections of this review, the conclusions drawn are much different when one considers investigations using conditioned place preference (CPP).
NE and Opiate SA
As with psychostimulant SA, research in the 1970s began to emphasize the importance of catecholamines in the mediation of opiate SA. A series of experiments demonstrated that depletion of NE and DA with AMPT, (which inhibits tyrosine hydroxylase) prevents or attenuates the SA of morphine in rodents (Davis and Smith, 1977) and in nonhuman primates (Pozuelo and Kerr, 1972). When treatments that could distinguish between the contributions of NE and DA were developed, researchers began to elucidate a role for NE in the mediation of morphine's behavioral effects. For example, reduction of NE synthesis with FLA-57, a dopamine β-hydroxylase (DBH) inhibitor, attenuates the oral intake of morphine in rats (Brown et al, 1978). Nonetheless, despite this demonstration, most research in the 1970s and 1980s focused on DA mediation of morphine reinforcement at the expense of NE, due in large part to the emerging DA hypothesis of psychostimulant SA.
Research into the DA hypothesis of opiate reinforcement has indeed indicated a role for this neurotransmitter, but experimental results have been fraught with inconsistencies (reviewed by Pierce and Kumaresan, 2006). For example, while 6-OHDA lesions do impair the acquisition of heroin SA (Singer and Wallace, 1984) and decrease morphine SA (Smith et al, 1985) in some studies, these results are not always replicated (Pettit et al, 1984; Dworkin et al, 1988, Gerrits and Van Ree, 1996). In addition, DA receptor antagonism does not consistently alter opiate SA behavior (Ettenberg et al, 1982; Van Ree and Ramsey, 1987; Gerber and Wise, 1989; Gerrits et al, 1994).
NE and Ethanol SA
Following the pattern established by psychostimulants and opiates, DA has been the main focus of research examining the neurochemical mediation of ethanol's reinforcing effects (Wise, 1980; Koob et al, 1998). Electrophysiological, pharmacological, and genetic experiments have established a clear role for DA in voluntary ethanol consumption (eg, Koob et al, 1994; El-Ghundi et al, 1998; Phillips et al, 1998). For example, SA of ethanol increases nucleus accumbens (Nac) DA release in rodents (Weiss et al, 1993, 1996; Gonzales and Weiss, 1998; Nurmi et al, 1998; Olive et al, 2000; Melendez et al, 2002; Hungund et al, 2003), and DA D1 and D2 receptor agonists and antagonists modulate ethanol SA in some circumstances (Weiss et al, 1990; Hubbell et al, 1991; Dyr et al, 1993; Rassnick et al, 1993a; Ng and George, 1994; Silvestre et al, 1996; Cohen et al, 1998, 1999; Boyce and Risinger, 2002; D’Souza et al, 2003; Zocchi et al, 2003). In addition, genetic deletion of D1 or D2 DA receptors decreases ethanol SA (El-Ghundi et al, 1998; Phillips et al, 1998; Risinger et al, 2000). However, inconsistencies similar to those obtained in examining the DA mediation of opiate reinforcement have plagued the field. For example, 6-OHDA lesions of the NAc do not alter ethanol SA in rats (Lyness and Smith, 1992; Rassnick et al, 1993b; Ikemoto et al, 1997, Koistinen et al, 2001). Further, there have been some conflicting results regarding the effects of pretreatment with dopaminergic agents (Goodwin et al, 1996; Silvestre et al, 1996).
For these reasons, alternate hypotheses of the neurochemistry of ethanol reinforcement have been postulated, including the suggestion that NE, and not DA, is the critical neurotransmitter (Amit and Brown, 1982). For example, acute administration of ethanol modulates the synthesis, turnover, and release of central NE (Corrodi et al, 1966; Carlsson and Lindqvist, 1973; Hunt and Majchrowicz, 1974; Pohorecky and Jaffe, 1975; Karoum et al, 1976), and the activity of noradrenergic neurons (Aston-Jones et al, 1982; Pohorecky and Brick, 1988; Verbanck et al, 1990). Moreover, ethanol has a greater effect on NE turnover and release than on DA (Corrodi et al, 1966; Hunt and Majchrowicz, 1974). Both chemical lesioning of the NE system and blocking NE synthesis via DBH inhibitors reduce voluntary ethanol intake, whereas DA lesions do not (Brown et al, 1977; Kiianmaa et al, 1979; Rassnick et al, 1993b).
Although these studies indicate an important role for NE in ethanol-mediated behaviors, other conflicting research has muddied the waters. Depending on the site of administration and the strain of rat used, chemical lesions of noradrenergic neurons with 6-OHDA can increase (Melchior and Myers, 1976; Kiianmaa, 1980), decrease (Melchior and Myers, 1976; Corcoran, Lewis, and Fibiger, 1983), or have no effect (Melchior and Myers, 1976; Richardson and Novakovski, 1978) on voluntary ethanol consumption. There are also conflicting data on DBH inhibitors (Amit et al, 1977; Daoust et al, 1990) and adrenergic agonists (Andreas et al, 1983; Grupp et al, 1989). Yet in spite of these conflicting results, most research has favored a prominent role for noradrenergic function in alcohol reward.
PART II: THE NORADRENERGIC THEORY OF DRUG REWARD: A RESURRECTION
NE and Psychostimulant-Induced Locomotion/Sensitization
Although not strictly a test of potential drug reward, measurement of locomotor activity has been critical to understanding molecules and pathways contributing to drug addiction for two key reasons. First, most drugs of abuse—including psychostimulants, opiates, and ethanol—produce locomotor hyperactivity, making this trait a generally reliable (albeit imperfect) predictor of abuse potential. Drug-induced locomotor hyperactivity is profoundly robust and reproducible, and the study of brain regions and signaling pathways controlling this phenomenon has significantly contributed to our understanding of how addictive drugs affect the brain. Second, drug-induced locomotor activity demonstrates sensitization, which may model progressive drug craving during the addiction process and manifests as increasing hyperactivity in response to the repeated administration of drugs of abuse (reviewed by Robinson and Berridge, 2000).
In sharp contrast to the paucity of SA data supporting the role of NE in psychostimulant addiction, another collection of studies has established that NE—acting primarily via α1-adrenergic receptors (α1ARs)—is essential for drug-induced locomotor activity and sensitization. For example, LC lesions attenuate amphetamine-induced locomotion (Mohammed et al, 1986). Also, administration of the α1AR antagonist prazosin, either systemically or directly into the prefrontal cortex (PFC), reduces both amphetamine- and cocaine-induced locomotion and sensitization (Snoddy and Tessel, 1985; Dickenson et al, 1988; Blanc et al, 1994; Darracq et al, 1998; Drouin et al, 2002; Weinshenker et al, 2002a; Wellman et al, 2002; Auclair et al, 2004; Salomon et al, 2006). Furthermore, α1bAR knockout mice are refractory to psychostimulant-induced locomotor activity and sensitization (Drouin et al, 2002; Auclair et al, 2004; Salomon et al, 2006). Conversely, elevating extracellular NE levels via blockade of α2AR inhibitory autoreceptors or via genetic ablation of the NE transporter (NET) increases the locomotor response to psychostimulants (Xu et al, 2000; Villégier et al, 2003). Although not as exhaustively investigated, α2- and βARs also appear to modulate this drug-induced behavior (Harris et al, 1996; Villégier et al, 2003). When combined, the results of these studies provide compelling evidence that NE is critical for psychostimulant-induced locomotor activity and sensitization.
NE and Psychostimulant CPP
CPP has recently been among the most popular measures of drug reward. In this paradigm, one set of contextual cues is paired with the drug of interest, whereas a different set of cues is paired with a vehicle control. After initial conditioning, an animal is then allowed unrestricted access to both contexts in the absence of drug, and an increase in time spent in the drug-paired context is interpreted as drug-associated reward. Although most drugs that support operant SA also support a CPP in rodents, there are some discrepancies, and each paradigm likely measures distinct reward processes. The primary differences involve active (SA) vs passive (CPP) drug administration, and operant responding for ‘immediate’ reward (SA) vs expression of a learned context-drug reward association in a drug-free state (CPP). Both paradigms have inherent advantages and disadvantages, and both have helped reveal molecules and circuits underlying drug reward (reviewed by Bardo and Bevins, 2000).
Surprisingly, there are few studies investigating the possible role of NE in psychostimulant CPP. Ventura et al (2003) found that a selective depletion of NE in the PFC abolishes amphetamine CPP in mice, whereas NET knockout mice with excess levels of extracellular NE show enhanced cocaine CPP. One caveat before drawing conclusions based on these results: NE is known to play an important role in some aspects of learning and memory. Because CPP is an associative learning paradigm, manipulations of NE could alter the development or expression of a psychostimulant CPP in the absence of any effect on the rewarding properties of the drugs. However, this is unlikely, as mice that completely lack NE still express a normal conditioned taste aversion to lithium chloride and ethanol and show a CPP to food (Weinshenker et al, 2000; Schank et al, 2006; Olson et al, 2006). Interestingly, NE depletion early in development either has no effect (neonatal 6-OHDA lesion; Spyraki et al, 1982a) or even enhances (DBH knockout mice; Schank et al, 2006) psychostimulant CPP, probably due to compensatory changes in the DA system during development (see below). NE may be an important mediator of the aspects of drug reward that are measured by the CPP paradigm, but clearly more work is required to define its influence, particularly with regard to the effects of adrenergic agonists and antagonists. This is an especially intriguing question—whereas DA is necessary for amphetamine CPP, it may not be necessary for cocaine CPP under all conditions (Spyraki et al, 1982a, 1982b; Miner et al, 1995; Baker et al, 1996; Sora et al, 1998; Tzschentke and Schmidt, 1998).
NE and Psychostimulant SA: A Reassessment
Owing to the absence of noradrenergic drug effects on SA (with the exception of clonidine), NE was simply written off as a potential mediator of psychostimulant reinforcement. Again, however, this conclusion was based on data examining a single phase of SA: the maintenance of a previously learned behavior. Primary reinforcement is only one facet of drug addiction, and perhaps not even the most important one, at least from a clinical standpoint. Current concepts in pharmacotherapy for drug dependence have aimed at preventing relapse, rather than disrupting primary reinforcement. The reinstatement phase of SA, during which noncontingent drug priming, drug-associated cues, or stress can trigger a previously extinct SA behavior, has become the standard paradigm for studying relapse (reviewed by Shaham et al, 2003).
In stark contrast to the lack of influence on maintenance of psychostimulant SA, the effects of noradrenergic drugs on reinstatement of cocaine and amphetamine drug seeking are profound and clear. NE was first implicated in reinstatement by Davis et al (1975). They found that DBH inhibitors that block NE synthesis attenuate reinstatement of amphetamine SA. The clearest case for noradrenergic involvement emerges in the stress-induced reinstatement paradigm. Systemic administration of clonidine or guanabenz, α2AR agonists that decrease NE release by activating inhibitory autoreceptors, attenuates footshock-induced reinstatement in rats (Erb et al, 2000). Furthermore, blockade of α2AR autoreceptors with either yohimbine or RS-79948 reinstates cocaine seeking in squirrel monkeys in the absence of any stressors (Lee et al, 2004). These effects likely involve stress-related circuitry in the extended amygdala, as local infusions of βAR antagonists in the bed nucleus of the stria terminalis (BNST) or in the central nucleus of the amygdala (CeA) also block footshock-induced reinstatement of cocaine SA in rats (Leri et al, 2002). Intriguingly, chronic cocaine SA in rhesus monkeys elevates NET density in the BNST to a greater extent than any reported changes in DAT, D1, or D2 receptors within the striatum of the same monkeys (Letchworth et al, 2001; Nader et al, 2002; Porrino et al, 2002; Macey et al, 2003). Given the known role of NE in central stress responses, these results indicate that NE release in the extended amygdala is required for stress-induced reinstatement. Again, while not necessarily pivotal for primary drug reinforcement, NE is certainly involved in another crucial aspect of addiction. Also of note here is the fact that the influence of NE on reinstatement is not limited to stress paradigms. Noncontigent injections of cocaine powerfully reinstate cocaine SA in normal rats, but not those pretreated with the α1AR antagonist prazosin (Zhang and Kosten, 2005). Because the βAR antagonists do not block cocaine-induced reinstatement, this would suggest that NE is critical for both stress- and drug-primed reinstatement, but via distinct receptors and pathways.
Finally, there are a few recent studies that have used new tools to reassess the role of the noradrenergic system in primary psychostimulant reinforcement. In a SA paradigm, NET knockout mice that have excess extracellular NE show a four-fold increase in their rate of cocaine intake, suggesting that chronic NET ablation causes a decrease in the reinforcing properties of cocaine (Rocha et al, 2003). Furthermore, while wild-type mice readily self-administer cocaine orally in a two-bottle free-choice paradigm, α1bAR knockout mice do not (Drouin et al, 2002). This is probably due to differences in drug reward sensation, not taste perception, as no genotype differences were observed for sucrose preference or quinine aversion. These results indicate that genetic alterations in noradrenergic pathways can modify the reinforcing properties of cocaine.
NE and Opiate-Induced Locomotion/Sensitization
As with the studies on the SA of psychostimulants, opiates, and ethanol, early experiments on morphine-induced locomotion highlighted the role of catecholamines in mediation of opiate-induced changes in locomotor activity. Inhibition of NE and DA synthesis with AMPT attenuates morphine-induced locomotion (Eidelberg and Schwartz, 1970; Davis et al, 1972; Buxbaum et al, 1973; Ayhan and Randrup, 1973). Subsequent research into the neurochemistry of morphine-induced locomotion revealed an important role for NE. Specific NE depletion by 6-OHDA lesions of the DNB potentiates the locomotor depressant and cataleptic effects of morphine in rats (Roberts et al, 1978). Moreover, pretreatment with FLA-63, a DBH inhibitor, reduces morphine-induced locomotion in rats (Ayhan and Randrup, 1973). In addition, the α-adrenergic antagonist, phenoxybenzamine, decreases the locomotion induced by morphine in mice (Estler, 1973) and rats (Ayhan and Randrup, 1973). Recent research has identified noradrenergic receptors within the PFC as being particularly important in subserving the locomotor effects of morphine. Infusions of prazosin, an α1-adrenergic antagonist, into this brain region attenuate the acute locomotor responses produced by morphine (Drouin et al, 2001).
Additional evidence for NE mediation of opiate-induced locomotion comes from genetic alterations of NE function. For example, morphine-induced locomotion is abolished in DBH knockout mice that lack NE, a deficit that is partially reversed by pharmacological restoration of NE or viral-mediated reexpression of DBH in the DNB or VNB (Olson et al, 2006). In addition, both genetic deletion and pharmacological blockade of α1bARs prevent morphine-induced locomotion (Drouin et al, 2002) and the development of locomotor sensitization (Auclair et al, 2004). Overall, the preponderance of available evidence indicates that NE is critical for the locomotor-activating effects of opiates.
NE and Opiate CPP
Similar to the pattern that emerged with studies examining the neurochemical substrates of behavioral actions associated with commonly abused drugs, early research on the neurochemistry of opiate CPP first examined the role of catecholamines as a group, then quickly came to focus on DA almost exclusively, once examination of NE function led to negative results. For example, in one of the earliest adaptations of the CPP paradigm (adapted from Beach, 1957, Schwartz and Marchok (1974) found that morphine approach responses to a Y-maze arm are attenuated by administration of the catecholamine inhibitor, AMPT. Further examination with haloperidol (a DA antagonist) and DETC (a DBH inhibitor) revealed that DA receptor blockade, but not NE inhibition, prevents morphine CPP. Similarly, both 6-OHDA lesions and haloperidol—but not neonatal 6-OHDA injections in rat pups (which leads to whole-brain depletion of NE)—attenuate heroin CPP (Spyraki et al, 1983). However, as mentioned previously, possible compensatory changes in the DA system during development could explain the lack of effect from the neonatal 6-OHDA injections. In fact, more recent work has indicated that DA is not required for opiate CPP. For example, mice utterly devoid of DA have surprisingly normal morphine CPP development, and simultaneous blockade of D1 and D2 receptors has no effect on the morphine CPP of drug-naïve animals (Laviolette et al, 2002; Hnasko et al, 2005). Together, these results suggest the existence of both DA-dependent and DA-independent pathways for opiate CPP.
Could the noradrenergic system be a part of the DA-independent pathway? Despite the relatively inauspicious debut of NE manipulations on opioid CPP, recent research indicates the necessity of noradrenergic function in the establishment of morphine CPP. For example, clonidine, an α2AR agonist, disrupts the establishment of a heroin CPP in rats, presumably by inhibiting NE release (Hand et al, 1989). In addition, both clonidine and prazosin (an α1AR antagonist) attenuate morphine CPP in mice, whereas yohimbine (another α2AR antagonist) increases morphine CPP (Zarrindast et al, 2002; Sahraei et al, 2004). Furthermore, both DBH knockout mice, which are incapable of producing NE, and α1bAR knockout mice fail to express a CPP over a wide range of morphine doses (Drouin et al, 2002; Olson et al, 2006). It is important to note in this context that morphine CPP can be restored in Dbh −/− mice by viral-mediated expression of DBH in the VNB (Olson et al, 2006). Finally, selective depletion of medial PFC noradrenergic afferents abolishes morphine CPP in mice (Ventura et al, 2005). Therefore, the present-day picture that is emerging indicates that NE is indeed required for the establishment of opiate CPP.
NE and Opiate SA: A Reassessment
A combination of several key factors suggests the need for a reexamination of NE's role in morphine SA: the inadequacies of the DA hypothesis of morphine reinforcement; the reduction of voluntary morphine intake following NE inhibition; and the importance of NE for morphine-induced locomotion and CPP. Moreover, the use of new tools (eg, subtype-specific noradrenergic agonists and antagonists, genetic manipulations) would provide a more accurate representation of NE's role in the mediation of opiate abuse. One study used knockout mice to reassess the role of the noradrenergic system in primary opiate reinforcement. Although wild-type mice readily self-administer morphine orally in a two-bottle free-choice paradigm, α1bAR knockout mice do not (Drouin et al, 2002). This result is probably due to differences in drug reward, rather than taste perception, as no genotype differences were observed for sucrose preference or quinine aversion.
As opposed to the relative dearth of information on NE mediation of morphine SA, the role of NE on reinstatement of opiate drug seeking—measured by either the reinstatement of drug SA or CPP—is emerging. NE was first implicated in reinstatement by Davis et al (1975), who found that DBH inhibitors attenuate the spontaneous reestablishment of opiate SA following a period of extinction. In addition, selective depletion of medial PFC noradrenergic afferents abolishes the reinstatement of an extinguished morphine CPP that has been produced by a priming injection of morphine (Ventura et al, 2005). Moreover, chronic treatment with venlaxfaxine, a dual NE/5-HT reuptake inhibitor, attenuates the reacquisition of a morphine CPP by a priming injection of morphine, whereas chronic treatment with the DA D2 antagonist, sulpiride, does not (Lu et al, 2001). These data indicate that morphine-seeking behavior induced by morphine abstinence or priming injections is at least partially mediated by NE.
In addition to relapse induced by morphine itself, stress-induced reinstatement of morphine SA also appears to be modulated by NE, as noted with psychostimulants. For example, both 6-OHDA lesions of the VNB and infusion of the α2AR agonist, clonidine, into the BNST block stress-induced reinstatement of morphine CPP (Wang et al, 2001). In addition, clonidine also prevents the stress-induced reinstatement of heroin-seeking behavior in rats (Shaham et al, 2000). Thus, NE appears to be critical for the stress-induced reinstatement of multiple classes of addictive drugs—a trend that extends even to ethanol, as we will see below.
NE and Opiate Withdrawal
The involvement of NE in opiate withdrawal has been exhaustively documented, and the reader is referred to several recent reviews (Nestler et al, 1994, 1999; Maldonado, 1997; Van Bockstaele et al, 2001). To avoid unnecessary redundancy, coverage of the subject will be omitted here.
NE and Ethanol-Induced Locomotion
Examination of ethanol-induced locomotion also identified the catecholamines as subserving this behavioral effect. For example, injection of AMPT blocks the locomotor stimulation produced by ethanol injection (Carlsson et al, 1972). In addition, depletion of forebrain NE with intracerebral injections of 6-OHDA exacerbates the locomotor suppressant effect of ethanol (Mason et al, 1979). Finally, administration of a moderate dose of ethanol produces an initial decrease, but a subsequent increase, in locomotor behavior. The initial locomotor decrease appears to be mediated by βARs, as the βAR antagonist, propranolol, selectively blocks the locomotor inhibition. The locomotor activation produced by ethanol later in the cycle, on the other hand, is blocked by phentolamine, an αAR antagonist (Matchett and Erickson, 1977).
NE and Ethanol CPP
There are no published studies examining the potential role of NE in the mediation of ethanol CPP.
NE and Ethanol SA: A Reassessment
New results reinforce the idea that NE contributes to the primary rewarding effects of ethanol. DBH knockout mice demonstrate a reduced voluntary ethanol consumption (Weinshenker et al, 2000), and while this reduction may result from a deficit in ethanol reward, it also may be due to the increased sensitivity of these mice to ethanol's aversive effects, such as sedation and hypothermia (Weinshenker et al, 2000). Another study showed that reducing NE transmission via activation of the α2AR autoreceptor with lofexidine attenuates alcohol SA, whereas increasing NE via blockade of this receptor enhances alcohol SA (Le et al, 2005). Finally, like stress-induced reinstatement of psychostimulants and opiates, relapse to ethanol-seeking behavior following footshock is attenuated by an α2AR agonist (Le et al, 2005).
PART III: DOPAMINE-DEPENDENT AND -INDEPENDENT MEDIATION OF DRUG ADDICTION BY NE
Although it is clear that NE signaling is important for at least some aspects of drug addiction, including primary reward, the underlying mechanisms have yet to be fully elucidated. In the present section we will discuss whether the effects of NE on responses to drugs of abuse depend on interactions with the DA system.
Control of Dopamine Neuron Firing and Dopamine Release by NE
Because DA is critical to many aspects of drug reward, understanding how NE influences DA signaling is of the utmost importance. The mesolimbic and mesocortical DA systems, comprised of projections from the ventral tegmental area (VTA) to the NAc and PFC, respectively, receive noradrenergic innervation and are modulated by NE. A wiring diagram of the functional interactions between the noradrenergic and dopaminergic systems is shown in Figure 1. Noradrenergic neurons from the LC, A1, and A2 nuclei innervate the VTA (Jones et al, 1977; Simon et al, 1979; Liprando et al, 2004) and provide excitatory drive to midbrain DA neurons. Electrical stimulation of the LC induces burst firing of VTA neurons, whereas burst firing is blocked by the α1adrenoreceptor (α1AR) antagonist, prazosin (Grenhoff et al, 1993; Grenhoff and Svensson, 1993). Conversely, lesions of the LC decrease striatal DA neuron activity (Tassin et al, 1979) and DA release (Russell et al, 1989; Lategan et al, 1990; Lategan et al, 1992). The PFC, a brain region implicated in responses to psychostimulants, also receives dense noradrenergic input from the LC (Swanson and Hartman, 1975; Morrison et al, 1981), which then sends excitatory glutamatergic projections to dopaminergic VTA neurons—though this connection may involve another glutamatergic relay nucleus (Carr and Sesack, 2000). Finally, the VNB projects directly to the NAc (Berridge et al, 1997; Delfs et al, 1998; Tong et al, 2006). These results form the basis of a functional connection between the noradrenergic and dopaminergic systems that is a likely component of central responses to drugs of abuse. Which (if any) of these circuits underlies the effects of NE on drug responses?

A wiring and neurotransmitter model for noradrenergic influence of psychostimulant responses. A1 and A2, brainstem noradrenergic cell groups; LC, locus coeruleus; PFC, prefrontal cortex; VTA, ventral tegmental area; NAc, nucleus accumbens; NE, norepinephrine; DA, dopamine; Glu, glutamate. Psychostimulant administration increases extracellular DA in the NAc and PFC and NE in the VTA, PFC, and NAc. NE signaling in the VTA induces burst firing of dopaminergic VTA neurons and increases DA release in the NAc. NE signaling in the PFC activates pyramidal neurons, which release Glu in the VTA resulting in increased excitability and more DA release in the NAc. Many of these noradrenergic inputs are mediated by the α1AR. The convergence of these signals in the NAc and PFC leads to psychostimulant-induced behaviors via downstream neuronal networks. Other cortical and subcortical inputs are also likely involved in the processes underlying addiction development, maintenance, and relapse.
For the psychostimulants, results from multiple studies have demonstrated that the LC-PFC projection is critical for DA release in the NAc. A selective lesion of PFC NE abolishes both amphetamine-induced (Ventura et al, 2003) and morphine-induced (Ventura et al, 2005) DA release in the mouse NAc. Again, the α1AR appears to be the primary mediator of this effect. α1Adrenoreceptor activation excites PFC pyramidal neurons (Marek and Aghajanian, 1999), and local infusions of prazosin (an α1AR antagonist) directly into the PFCs of rats blocks ‘functional’ DA release in the NAc (ie, the DA release associated with a behavioral response; Blanc et al, 1994; Darracq et al, 1998). Amphetamine-induced DA release in the NAc is also abolished in α1bAR knockout mice and in DBH knockout mice that lack NE (Auclair et al, 2002; Schank et al, 2006). Because the treatments that attenuate DA release in the NAc also attenuate psychostimulant-induced locomotion and CPP, the behavioral responses to psychostimulants appear to depend on NE activation of α1ARs in the PFC, which in turn promotes DA neuron firing and DA release in the ventral striatum.
The importance of the direct noradrenergic projections to the VTA and NAc has not been extensively studied, at least not directly (eg, by depleting NE or infusing AR antagonists locally into these regions and assessing drug reward, locomotion, etc). Nonetheless, there are some indications that these pathways are also important. Infusion of either DA or NE directly into the NAc stimulates locomotor activity in the rat, whereas local infusion of haloperidol attenuates the effects of both catecholamines (Pijnenburg et al, 1975; Svensson and Ahlenius, 1982). Furthermore, infusion of the α1AR antagonist, prazosin, into the NAc attenuates DA release in rats (Sommermeyer et al, 1995) and reduces the locomotor activity of mice in a novel environment (Stone et al, 2004). Although α1ARs are detectable in the NAc using radioligand binding, α1AR mRNA has not been found in accumbal neurons (Rainbow and Biegon, 1983; Day et al, 1997; Domyancic and Morilak, 1997). This somewhat limited set of results suggests that NE facilitates DA release and locomotor activity via α1ARs located on DA neuron terminals. Blockade of βARs with propranolol was reported to increase accumbal DA release and cocaine-induced locomotor activity, although the propranolol was administered peripherally, therefore the anatomical localization of these effects could not be pinpointed (Harris et al, 1996). In slice experiments, Nicola and Malenka (1998) reported that, like DA, NE depresses excitatory postsynaptic potentials in the NAc via an αAR-dependent mechanism. Although they did not ‘localize’ this effect, their data suggest that NE influences the activity of accumbal neurons directly. Because they used a nonselective αAR blocker (phentolamine), and as accumbal neurons do not express α1AR mRNA, the depression of EPSPs observed in this study is likely mediated by the α2AR, which is highly expressed in the striatum. The effects of accumbal infusion of adrenergic agonists/antagonists on drug-induced DA release and behavior have not been assessed. It would also appear that there is some reciprocal modulation of accumbal NE release by DA. Vanderschuren et al (1999) reported that D1 receptor activation facilitates, whereas D2 receptor activation depresses, accumbal NE release in slice culture.
The data on direct noradrenergic modulation of VTA DA neurons are also somewhat difficult to interpret, but quite enticing. Although much of the work has focused on α1AR modulation of VTA DA neurons’ firing properties, neither radioligand binding nor mRNA in situ hybridization techniques has reliably detected α1ARs in the VTA (Jones et al, 1985; Palacios et al, 1987; Pieribone et al, 1994). Thus, it is important to bear in mind that, until more sensitive techniques (eg, single-cell RT-PCR, electron microscopy) are employed to identify the exact anatomical localization of α1ARs in the VTA, many of the following conclusions remain speculative. In the mid-1990s, Grenhoff et al published a landmark series of studies examining modulation of DA cells by NE. In anesthetized rats, stimulation of the LC increases burst firing of VTA DA neurons, whereas decreasing NE release with an α2AR agonist or blocking α1ARs with prazosin will eliminate bursting and regularize the firing of these cells (Grenhoff and Svensson, 1989, 1993; Grenhoff et al, 1993). However, because the noradrenergic drugs were administered systemically, their effects could not be localized. The researchers went on to reexamine these effects in slice culture and found that α1AR activation has two effects—it both depolarizes DA cells and increases the frequencies of inhibitory postsynaptic potentials. The former effect was localized to DA neurons, because blocking synaptic transmission with tetrodotoxin (TTX) had no impact, whereas the latter effect was TTX sensitive and was attributed to the excitation of interneurons resulting in GABA release onto DA neurons (Grenhoff et al, 1995). This work has been ongoing the past few years and was eventually extended to include the effects of amphetamine on DA neuron activity. As with previous reports, Shi et al (2000) found that amphetamine depresses VTA DA neuron firing in vivo, primarily via D2 autoreceptor activation. However, when D2 receptors are blocked, the inhibitory effects of amphetamine switch to an α1AR-mediated excitation of these cells (increased firing rate and bursting). They went on to show that, even in the absence of D2 blockade, α1AR activation is responsible for the increase in the psychostimulant-induced augmentation of the slow oscillation firing pattern in VTA cells (Zhou et al, 2006). Again, as prazosin was administered peripherally in these experiments, localization of the α1AR effect could not be determined. Some of the most compelling data indicating a direct NE effect on VTA DA neurons is that, in slice culture, activation of α1ARs increases amphetamine-induced DA neuron activity by counteracting the inhibitory effect of mGluR1 activation (Paladini et al, 2001). This effect is likely due to direct action on DA neurons, both because the noradrenergic drugs were applied iontophoretically rather than superfused in the bath, and because the effects persisted in the presence of TTX and in the absence of Ca2+. Interestingly, Paladini et al also identified a direct, α1AR-mediated inhibitory effect on DA neurons (Paladini and Williams, 2004). Thus, it would appear that α1AR activation excites as well as inhibits midbrain DA neurons. In the absence of pharmacological intervention, these cells typically have a bimodal spontaneous firing pattern: bursts—which greatly enhance DA release in the NAc—followed by periods of quiescence, when DA release is low or absent, and high variability in the interspike interval (Gonon, 1988; Bean and Roth, 1991; Grenhoff and Svensson, 1993). When noradrenergic transmission is blocked, both the excitatory and inhibitory effects of NE vanish, resulting in a regularization of DA neuron firing, and overall decreased DA utilization (Anden et al, 1970; Anden and Grabowska, 1976; Grenhoff and Svensson, 1989, 1993; Zhou et al, 2006). This regularization is critical because, as Tassin, Puglisi-Allegra, and others have shown, regularized DA release in the NAc is not functional in a behavioral sense; ‘functional’ DA release, which is characterized by the highly variable burst-quiescence pattern that is correlated with behavioral change, is largely dependent on α1AR activation (Darracq et al, 1998; Auclair et al, 2002). These results could explain why genetic or pharmacologic blockade of α1AR signaling attenuates amphetamine-induced DA release in the NAc and behavioral responses to psychostimulants. We are proposing a model in which fine tuning of DA neuron firing and accumbal DA release by combined NE signaling in the VTA, NAc, and PFC is critical for psychostimulant-associated behaviors.
Paradoxical Hyperdopaminergic State Following Chronic NE Depletion: Lessons from Dopamine β-Hydroxylase Knockout Mice
In the preceding sections, we reviewed literature demonstrating that NE, primarily via α1AR signaling, is required for proper DA neuron firing, DA release in the NAc, and psychostimulant-induced locomotion, sensitization, and CPP. These studies all shared a common feature: the blockade of NE signaling that led to depression of DA transmission and attenuation of psychostimulant-induced behaviors was either acute (eg, single exposure to prazosin) or involved just a single receptor subtype (eg, α1b knockout mice). We initiated a series of studies that aimed to elucidate the effects of chronic NE depletion on DA transmission and psychostimulant responses by using DBH knockout (Dbh −/−) mice, which lack NE completely from birth (Thomas et al, 1995; 1998). Understanding the effects of chronic NE depletion is important for several reasons. First, drug addiction is a chronic disease. Second, the proclivity to drug addiction is influenced by genetic variation that is in play throughout development and adulthood. Finally, any pharmacological intervention used to treat addiction will be chronic of necessity.
We predicted that the behavior of Dbh −/− mice would mirror that of prazosin-treated mice or α1bAR knockout mice. Surprisingly, instead of an attenuated response to psychostimulants, Dbh −/− mice are hypersensitive to the locomotor, rewarding, and aversive effects of cocaine and amphetamine (Weinshenker et al, 2002a; Schank et al, 2006). To explain the underlying mechanisms of this paradoxical hypersensitivity to psychostimulants on the part of Dbh −/− mice, we examined DA transmission by microdialysis and radioligand binding. We found that Dbh −/− mice have a reduction in basal extracellular DA in the NAc and CP, whereas amphetamine-induced DA release is abolished in the NAc and attenuated in the CP and PFC (Schank et al, 2006; Seeman et al, 2005). It is important to note that the ‘ectopic’ DA produced in ‘noradrenergic’ neurons in Dbh −/− mice is not the cause of the hypersensitivity; although Dbh −/− mice indeed produce more DA in brain tissue than normal mice (Thomas et al, 1998; Bourdelat-Parks et al, 2005), overall DA release is hampered. These results are consistent with those discussed in the previous section demonstrating that NE is important for DA neuron activity and DA release. It has been well established that the DA system compensates for a loss of dopaminergic tone by upregulating DA receptor signaling capacity, and that is exactly what happens in Dbh −/− mice. Using radioligand binding in the presence and absence of guanine nucleotide, which can discriminate between low- and high-affinity state DA receptors, we found that the density of high-affinity state DA receptors is increased three to to six-fold in the NAc and CP of Dbh −/− mice (Seeman et al, 2005; Schank et al, 2006). Furthermore, Dbh −/− mice are behaviorally hypersensitive to quinpirole, a direct D2 agonist (Weinshenker et al, 2002a). Thus, when NE is blocked acutely, DA transmission and behavioral responses to psychostimulants appear to be attenuated. In contrast, when NE is blocked chronically, a similar attenuation in DA release occurs, but over time the DA system compensates by upregulating high-affinity state postsynaptic DA receptors, which results in a behavioral hypersensitivity to psychostimulants. In support of this hypothesis, other treatments that result in long-term NE depletion (eg, neurotoxic lesions of the LC or chronic treatment with a DBH inhibitor) also produce hypersensitivity to psychostimulants and direct DA agonists (Donaldson et al, 1976; Lategan et al, 1990; Harro et al, 2000). We will revisit this model when we discuss the current and potential uses of DBH inhibitors for treating cocaine dependence.
Nontraditional NE–DA Interactions
Besides the important influence of NE transmission on DA signaling, the noradrenergic system can influence the dopaminergic system in at least three ‘nontraditional’ ways: uptake of DA by noradrenergic neurons, release of DA by noradrenergic neurons, and activation of adrenergic receptors by DA.
The plasma membrane NET, which is responsible for the uptake of extracellular NE, is also capable of transporting DA in vitro (Raiteri et al, 1977). This raises the possibility that the NET may be at least partially responsible for DA uptake in brain regions innervated by both DA and NE neurons. This is an especially intriguing hypothesis, because the ability of selective DAT blockers to increase extracellular DA concentrations is oddly low in certain brain regions, such as the PFC (Carboni et al, 1990; Di Chiara et al, 1992). Indeed, further studies by a number of groups using selective transporter blockers and transporter knockout mice confirmed that the NET is primarily responsible for DA uptake in the PFC, where DAT is scarce, and significantly contributes to DA uptake in regions where NET and DAT coexist, such as the NAc shell and BNST (Mundorf et al, 2001; Moron et al, 2002; reviewed by Carboni and Silvagni, 2004). These results indicate that the psychostimulant-induced increase in extracellular DA in the PFC is mediated predominantly by NET and not DAT blockade. In addition, when DAT is impaired (eg, DAT knockout mice), the NET may represent the primary mechanism for DA uptake in other brain regions. Interestingly, nisoxetine, a selective NET blocker, cannot support a CPP in normal mice, but it can in DAT heterozygote and knockout mice (reviewed by Uhl et al, 2002).
DA is synthesized by all NE neurons and is converted to NE by DBH during NE biosynthesis. Under normal conditions, tyrosine hydroxylase—not DBH—is rate limiting for NE synthesis (reviewed by Udenfriend, 1966). However, the conversion of DA to NE may not be completely efficient, and DBH can become rate limiting when neuronal activity is high or when DBH is genetically compromised or pharmacologically inhibited. Under these conditions (and perhaps others) ‘noradrenergic’ neurons do appear to release DA (Scatton et al, 1984; Devoto et al, 2001; Weinshenker et al, 2002b; Carboni and Silvagni, 2004; Bourdelat-Parks et al, 2005; Devoto et al, 2005).
Because DA is structurally similar to NE, DA can activate cloned ARs in cell culture, although with less potency than NE by 2–3 orders of magnitude (eg, Zhang et al, 2004). Paladini et al, 2001 showed that the ability of NE to activate α1ARs in VTA neurons in brain slices is recapitulated by DA. Debate has swirled for years around the issue of whether DA can activate α1ARs—although some evidence exists in the affirmative, alternative explanations can often undercut its persuasiveness. In addition, work with Dbh −/− mice has indicated that DA cannot activate α1ARs in vivo. For example, if DA could activate α1ARs, Dbh −/− mice would be expected to: lack phenotypes consistent with the absence of α1ARs; lack phenotypes that could be rescued by an α1AR agonist; and respond to α1AR antagonists. On the contrary, Dbh −/− mice do have reductions in DA release consistent with an absence of α1ARs (Schank et al, 2006); they also have a seizure susceptibility phenotype that is rescued by an α1AR agonist (Weinshenker et al, 2001) and are completely indifferent to the α1AR antagonist, prazosin (Weinshenker et al, 2002a). Currently, activation of an AR by DA in vivo is supported by good evidence in just one instance: α2cARs in the striatum. Given the relatively sparse noradrenergic innervation of the striatum (Lindvall and Björklund, 1974; Swanson and Hartman, 1975), there is a surprising abundance of α2ARs in this region, especially the α2cAR (Ordway et al, 1993; Nicholas et al, 1993; Uhlen et al, 1997). This paradox led Ordway et al to propose that DA, rather than NE, is the endogenous ligand for α2cARs in the striatum (Zhang et al, 1999). They showed that activation of cloned α2cARs with either NE or DA inhibits forskolin-induced cAMP accumulation (Zhang et al, 1999) and that α2cAR activation inhibits GABA release in striatal slices (Zhang and Ordway, 2003). Thus, DA activation of these ‘adrenergic’ receptors may be a normal component of striatal DA signaling and could modulate responses to drugs of abuse.
Some Effects of NE on Drug Responses may be Independent of DA
Although the effects of noradrenergic manipulations on psychostimulant responses appear to be primarily mediated by the modulation of DA release, some of the mechanisms by which NE influences opiate and ethanol reward may be independent of DA. The criteria for this putative DA-independent pathway are that manipulations of NE function alter drug responses, whereas DA manipulations are without consistent effect. For example, Dbh −/− and α1bAR knockout mice do not develop morphine CPPs, indicating that normal NE function is critical for opiate-conditioned reward (Olson et al, 2006; Drouin et al, 2002), whereas DA-deficient mice display normal CPP acquisition (Hnasko et al, 2005). In addition, intra-NAc infusion of a broad-spectrum DA antagonist has no effect on morphine CPP in drug-naïve animals (Laviolette et al, 2002). A possible anatomical substrate for these potential DA-independent effects is the direct NE projection of the VNB to the NAc (Berridge et al, 1997; Delfs et al, 1998; Tong et al, 2006), a hypothesis buttressed by the fact that 6-OHDA lesions of accumbal DA (NE content was unaffected in this particular study) selectively attenuate cocaine SA, but not heroin SA, in rats (Pettit et al, 1984), whereas kainic acid lesions of the same structure disrupt both cocaine and heroin SA (Zito et al, 1985). Together, these data suggest that other neurotransmitters in the NAc, such as NE, mediate the primary reinforcing effects of opiates. Other potential neuroanatomical candidates are the PFC, amygdala, and BNST, which receive direct noradrenergic input and modulate various aspects of drug reward and addiction. One important caveat—the negative DA findings are inconsistent with other reports demonstrating that DA antagonism prevents opioid CPPs and SA (reviewed by Bardo and Bevins, 2000; McBride et al, 1999; Tzschentke, 1998; Bardo, 1998). However, these studies are complicated by the aversive and memory-impairing properties of DA receptor antagonists themselves.
Noradrenergic manipulations that alter ethanol intake may also occur independently of DA function. For example, chemical lesions of the NE system, blocking NE synthesis via DBH inhibitors, or genetic deletion of DBH will reduce voluntary ethanol intake, whereas DA lesions do not (Brown et al, 1977; Kiianmaa et al, 1979; Rassnick et al, 1993b; Weinshenker et al, 2000). Again, evidence does suggest that pharmacologic or genetic manipulation of D1 and D2 receptors can modulate ethanol SA in some circumstances (Weiss et al, 1990; Hubbell et al, 1991; Dyr et al, 1993; Rassnick et al, 1993a; Ng and George, 1994; Silvestre et al, 1996; Cohen et al, 1998, 1999; El-Ghundi et al, 1998; Phillips et al, 1998; Risinger et al, 2000; Boyce and Risinger, 2002; D’Souza et al, 2003; Zocchi et al, 2003). Further research teasing out the specific mechanisms of noradrenergic modulation of opioid and ethanol reward will be required to distinguish between DA-dependent and DA-independent pathways.
PART IV: INFLUENCE OF NORADRENERGIC GENE POLYMORPHISMS AND NORADRENERGIC PHARMACOTHERAPY ON DRUG DEPENDENCE
Addiction researchers have invested tremendous effort over the years to develop pharmacotherapies for drug dependence. In general, these potential therapies act by one of the following means: reducing drug reward, increasing drug aversion, or partially substituting for the abused drug. Results for each strategy have been mixed, ranging from substantial success (eg, methadone/buprenorphine maintenance for opiate dependence) to general failure (eg, DA agonists/antagonists for cocaine dependence). In this section, we will review the efficacy of noradrenergic compounds in the treatment of drug addiction.
Disulfiram and Cocaine Dependence
At the present time, disulfiram has shown probably the greatest promise of any compound for the treatment of cocaine addiction. Disulfiram (Antabuse®) has been used for over 50 years in the treatment of alcoholism (Fuller et al, 1986). Disulfiram inhibits the enzyme aldehyde dehydrogenase, which results in the accumulation of acetaldehyde, a toxic metabolic intermediate, upon ethanol ingestion. Acetaldehyde produces ‘the Antabuse reaction’, an aversive syndrome characterized by flushing, nausea, and vomiting. The desire to avoid this syndrome by reducing alcohol intake is believed to be responsible for the reductions in alcohol use in dependent individuals.
The idea of using disulfiram to treat cocaine addiction originates from the remarkable degree of comorbidity seen with alcohol dependence and cocaine dependence (Regier et al, 1990; Carroll et al, 1993; Higgins et al, 1993). Although preliminary findings indeed supported the efficacy of disulfiram in cocaine/alcohol codependent individuals (Carroll et al, 1998, 2000), results from three recent studies strongly suggest that comorbid alcohol use is not necessary for disulfiram treatment of cocaine dependence—in fact, nonalcohol-dependent subjects may derive even more benefit from disulfiram treatment than those who also abuse alcohol (George et al, 2000; Petrakis et al, 2000; Carroll et al, 2004). Because the drug combination of disulfiram and cocaine in the absence of alcohol does not result in acetaldehyde accumulation, the reduction of cocaine use with disulfiram treatment must depend on an interaction other than inhibition of aldehyde dehydrogenase.
As it happens, disulfiram is also a potent inhibitor of DBH. Most inhibitors of DBH, including disulfiram, chelate copper, thus depriving DBH of its required cofactor (Goldstein et al, 1964). Disulfiram inhibits DBH in animals, decreasing NE and increasing DA in both peripheral and central tissues (Musacchio et al, 1966; Karamanakos et al, 2001). In humans, disulfiram decreases NE and its metabolites in urine, blood, and cerebrospinal fluid (Takahashi and Gjessing, 1972; Major et al, 1979; Rogers et al, 1979; Hoeldtke and Stetson, 1980; Rosen and Lobo, 1987; Paradisi et al, 1991). Disulfiram inhibits DBH and aldehyde dehydrogenase similarly, with IC50 values in the low (μM) range for both enzymes (Green, 1964; Mays et al, 1998). Because the rewarding and aversive effects of cocaine are primarily mediated by NE and DA, we (along with others) have proposed that DBH inhibition is likely a key to the success of disulfiram treatment for cocaine dependence (McCance-Katz et al, 1998a, 1998b; George et al, 2000; Petrakis et al, 2000; Bourdelat-Parks et al, 2005; Schank et al, 2006; Sofuoglu and Kosten, 2006).
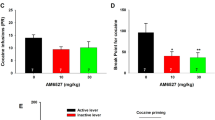
Because the treatment of alcoholism with disulfiram appears to depend on the drug's ability to create an aversive reaction to alcohol ingestion, a similar mechanism, mediated by DBH inhibition, may be responsible for its effect on cocaine dependence. But is there evidence to support such a mechanism of action? First, we point out that Dbh −/− mice have altered responses to cocaine. Perhaps of greatest significance, Dbh −/− mice display a conditioned place aversion to cocaine at doses that normally support a CPP in regular mice (Schank et al, 2006). In humans, a common polymorphism (allele frequency of 0.22) that accounts for much of the genetic variance in DBH activity was identified in the promoter region of the human DBH gene (C–T change at nucleotide position −1021) (Zabetian et al, 2001). CT heterozygotes have levels about 50% lower than those found in CC homozygotes, whereas TT homozygotes have very low DBH activity (∼10% of CC). Also of note, individuals receiving disulfiram treatment have reported a higher incidence of paranoia associated with cocaine use than those not receiving disulfiram (Hameedi et al, 1995; McCance-Katz et al, 1998a, 1998b), and individuals with genetically low DBH activity appear to be particularly susceptible to cocaine-induced paranoia (Cubells et al, 2000; R Malison, personal communication). Finally, there appears to be a pharmacogenetic interaction between disulfiram and DBH genotype. The effects of disulfiram and DBH mutations on catecholamine levels are additive in mice (Bourdelat-Parks et al, 2005), and individuals with low DBH activity are more susceptible to some aversive side effects of disulfiram, including psychosis (Heath et al, 1965; Ewing et al, 1977; Major et al, 1979) and sedation (Ewing et al, 1978). In a recent genotype-controlled pilot study, disulfiram effectively reduced cocaine intake only in individuals carrying at least one low-activity DBH allele (Cubells et al, 2000). Together, these combined results suggest the existence of a second aversive ‘Antabuse reaction’ that promotes cocaine abstinence and that is mediated by inhibition of DBH, not aldehyde dehydrogenase. Because blockade of NE signaling attenuates footshock-induced and cocaine-primed reinstatement of cocaine seeking in rats, an attenuation of factors that cause relapse, such as stress or drug exposure, could also contribute to the success of disulfiram. The next step, clearly, is to test the efficacy of selective DBH inhibitors in preclinical models of cocaine addiction and in cocaine-dependent individuals, preferably in a manner that will take DBH genotype into account. The possibility that disulfiram and other DBH inhibitors may be effective in treating dependence to other psychostimulants, such as methamphetamine, also merits exploration.
Other Noradrenergic Compounds Affecting Drug Abuse
Although disulfiram is the most promising noradrenergic treatment for drug dependence, there are others that have met with some success. NET blockers, such as desipramine and reboxetine, have been modestly effective in treating cocaine addiction (Kosten et al, 2005; McDowell et al, 2005; Szerman et al, 2005), although this may have more to do with their antidepressant activity than with a direct effect on cocaine responses, per se. One recent set of studies reported that the wake-promoting drug, modafinil, which is approved as a treatment for narcolepsy, was able to reduce cocaine intake in a dependent cohort (Dackis et al, 2003; 2005). Intriguingly, modafinil inhibits both DAT and NET, and both its locomotor and wake-promoting effects are attenuated by α1AR blockade in rodents and nonhuman primates (Duteil et al, 1990; Hermant et al, 1991; Stone et al, 2002; Madras et al, 2006). Perhaps its efficacy in treating cocaine dependence is also tied to α1AR signaling, although influences on glutamatergic transmission are another possibility (Ferraro et al, 1999; Dackis and O’Brien, 2003).
There were a few reports from the 1970s indicating that propranolol, a βAR antagonist, could block the euphoric effects of opiates and might be effective in treating opiate dependence in human addicts (Grosz, 1972). Moreover, subsequent preclinical research suggested that propranolol could attenuate some of the behavioral effects of morphine in rats (Black and Grosz, 1974; Black et al, 1975). However intriguing, the importance of this line of research was called into question, as subsequent studies were unable to replicate the original claim that propranolol could attenuate the euphoric effects of opiates in humans, and the issue has not been taken up again (Jacob et al, 1975; Resnick et al, 1976).
DBH and Alcoholism
As described previously, disulfiram inhibits aldehyde dehydrogenase, which results in the accumulation of a toxic metabolic intermediate—acetaldehyde—upon ethanol ingestion. Acetaldehyde produces the aversive symptoms that are presumed responsible for reductions in ethanol intake. Because disulfiram inhibits DBH and aldehyde dehydrogenase to a similar degree (Green, 1964; Mays et al, 1998), it is possible that disulfiram inhibition of DBH may be partly responsible for the reduction of ethanol ingestion following disulfiram administration. Amit et al (1976) examined this possibility by determining the efficacy of calcium carbimide, FLA-63, and disulfiram in decreasing ethanol intake and the effect each of these compounds had on acetylaldehyde levels following ethanol injection. Of the three compounds tested, calcium carbimide had the greatest effect on acetylaldehyde levels following ethanol injection, but the least effect on ethanol intake. Administration of disulfiram and FLA-63 (both DBH inhibitors) significantly reduced ethanol intake. Of the two compounds, FLA-63 had the least effect on acetylaldehyde levels, but was the most potent suppressor of ethanol intake. This series of results led the authors to conclude that disruption of NE synthesis may significantly contribute to the efficacy of disulfiram in the treatment of alcoholism.
DBH function has also been implicated in human alcoholism. Plasma DBH activity in alcoholic subjects is lower than that seen in nonalcoholics (Kohnke et al, 2002), and alcoholics have an increased frequency of the low-activity A allele of the DBH(*) 444G/A polymorphism (Kohnke et al, 2006). These studies indicate that the genetic determinants of DBH function may be associated with alcoholism. This is why pharmacological remediation with NE modulators may represent a rational therapeutic treatment strategy for alcohol abuse.
CONCLUSION
Although NE was dismissed as a key player in reward and addiction during the mid-1970s, more recent work has given us pause and led us to rethink the importance of this neurotransmitter. As always, it is a marvel how similar data can be perceived and interpreted so differently. When researchers discovered that DA is dispensable for opiate and ethanol SA and CPP for a number of addictive compounds, the general conclusion was that there had to be both DA-dependent and DA-independent reward pathways. In marked contrast, when NE was deemed unimportant for the maintenance phase of psychostimulant SA, it was dismissed as having no role in addiction at all—this despite the fact that NE does appear to be necessary for voluntary ethanol consumption, CPP for psychostimulants and opiates, and stress-induced reinstatement of multiple drugs of abuse. We propose that, as with DA, there are both NE-dependent and NE-independent circuits that influence drug response and addiction parameters. Further, some of the NE-dependent pathways (like those related to psychostimulant-induced behaviors) are mediated by the modulation of DA release, whereas others (eg, ethanol SA, opiate CPP) may not be.
To obtain a comprehensive picture of the role NE actually plays in drug reward and addiction, a number of important questions need to be addressed. What is the anatomical location for the influence of NE on ethanol reward? Is NE important for opiate SA? What is the exact role of NE release in the NAc and VTA in drug-induced behaviors? Do VTA neurons express adrenergic receptors (especially α1ARs), and if so, what types? Do functional polymorphisms in DBH and other genes in the noradrenergic pathway modulate aspects of addiction? Is the mechanism of disulfiram-induced cocaine abstinence really DBH inhibition? Finding answers to these questions will enhance our knowledge of reward and addiction pathways and may lead to novel treatments for drug dependence.
References
Amaral DG, Routtenberg A (1975). Locus coeruleus and intracranial self-stimulation: a cautionary note. Behav Biol 13: 331–338.
Amit Z, Brown ZW (1982). Actions of drugs of abuse on brain reward systems: a reconsideration with specific attention to alcohol. Pharmacol Biochem Behav 17: 233–238.
Amit Z, Brown ZW, Levitan DE, Ogren SO (1977). Noradrenergic mediation of the positive reinforcing properties of ethanol: I. Suppression of ethanol consumption in laboratory rats following dopamine-beta-hydroxylase inhibition. Arch Int Pharmacodyn Ther 230: 65–75.
Amit Z, Levitan DE, Lindros KO (1976). Suppression of ethanol intake following administration of dopamine-beta-hydroxylase inhibitors in rats. Arch Int Pharmacodyn Ther 223: 114–119.
Anden N, Grabowska M (1976). Pharmacological evidence for a stimulation of dopamine neurons by noradrenaline neurons in the brain. Eur J Pharmacol 39: 275–282.
Anden NE, Butcher SG, Corrodi H, Fuxe K, Ungerstedt U (1970). Receptor activity and turnover of dopamine and noradrenaline after neuroleptics. Eur J Pharmacol 11: 303–314.
Andreas K, Fischer HD, Schmidt J (1983). Effect of central effective substances on alcohol preference. Biomed Biochim Acta 42: 391–398.
Aston-Jones G, Cohen JD (2005). An integrative theory of locus coeruleus–norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28: 403–450.
Aston-Jones G, Foote SL, Bloom FE (1982). Low doses of ethanol disrupt sensory responses of brain noradrenergic neurones. Nature 296: 857–860.
Auclair A, Cotecchia S, Glowinski J, Tassin JP (2002). D-Amphetamine fails to increase extracellular dopamine levels in mice lacking alpha 1b-adrenergic receptors: relationship between functional and nonfunctional dopamine release. J Neurosci 22: 9150–9154.
Auclair A, Drouin C, Cotecchia S, Glowinski J, Tassin JP (2004). 5-HT2A and alpha1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur J Neurosci 20: 3073–3084.
Ayhan IH, Randrup A (1973). Behavioural and pharmacological studies on morphine-induced excitation of rats. Possible relation to brain catecholamines. Psychopharmacologia 29: 317–328.
Baker DA, Khroyan TV, O’Dell LE, Fuchs RA, Neisewander JL (1996). Differential effects of intra-accumbens sulpiride on cocaine-induced locomotion and conditioned place preference. J Pharmacol Exp Ther 279: 392–401.
Bardo MT (1998). Neuropharmacological mechanisms of drug reward: beyond dopamine in the nucleus accumbens. Crit Rev Neurobiol 12: 37–67.
Bardo MT, Bevins RA (2000). Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berlin) 153: 31–43.
Beach HD (1957). Morphine addiction in rats. Can J Psychol 11: 104–112.
Bean AJ, Roth RH (1991). Extracellular dopamine and neurotensin in rat prefrontal cortex in vivo: effects of median forebrain bundle stimulation frequency, stimulation pattern, and dopamine autoreceptors. J Neurosci 11: 2694–2702.
Berridge CW, Stratford TL, Foote SL, Kelley AE (1997). Distribution of dopamine beta-hydroxylase-like immunoreactive fibers within the shell subregion of the nucleus accumbens. Synapse 27: 230–241.
Berridge CW, Waterhouse BD (2003). The locus coeruleus–noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 42: 33–84.
Black WC, Grosz HJ (1974). Propranolol antagonism of morphine-influenced behavior. Brain Res 65: 362–367.
Black WC, McBride WJ, Grosz HJ (1975). Propranolol, 14C-morphine accumulation and avoidance: peripheral and central variables. Pharmacol Biochem Behav 3: 701–704.
Blanc G, Trovero F, Vezina P, Herve D, Godeheu AM, Glowinski J et al (1994). Blockade of prefronto-cortical alpha 1-adrenergic receptors prevents locomotor hyperactivity induced by subcortical D-amphetamine injection. Eur J Neurosci 6: 293–298.
Bourdelat-Parks BN, Anderson GM, Donaldson ZR, Weiss JM, Bonsall RW, Emery MS et al (2005). Effects of dopamine beta-hydroxylase genotype and disulfiram inhibition on catecholamine homeostasis in mice. Psychopharmacology (Berlin) 183: 72–80.
Boyce JM, Risinger FO (2002). Dopamine D3 receptor antagonist effects on the motivational effects of ethanol. Alcohol 28: 47–55.
Brown ZW, Amit Z, Levitan DE, Ogren SO, Sutherland EA (1977). Noradrenergic mediation of the positive reinforcing properties of ethanol: II. Extinction of ethanol-drinking behavior in laboratory rats by inhibition of dopamine-beta-hydroxylase. Implications for treatment procedures in human alcoholics. Arch Int Pharmacodyn Ther 230: 76–82.
Brown ZW, Amit Z, Sinyor D, Rockman GE, Ogren SO (1978). Suppression of voluntary ingestion of morphine by inhibition of dopamine-beta-hydroxylase. Arch Int Pharmacodyn Ther 232: 102–110.
Buxbaum DM, Yarbrough GG, Carter ME (1973). Biogenic amines and narcotic effects. I. Modification of morphine-induced analgesia and motor activity after alteration of cerebral amine levels. J Pharmacol Exp Ther 185: 317–327.
Carboni E, Silvagni A (2004). Dopamine reuptake by norepinephrine neurons: exception or rule? Crit Rev Neurobiol 16: 121–128.
Carboni E, Tanda GL, Frau R, Di Chiara G (1990). Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J Neurochem 55: 1067–1070.
Carlsson A, Engel J, Svensson TH (1972). Inhibition of ethanol-induced excitation in mice and rats by α-methyl-p-tyrosine. Psychopharmacologia 26: 307–312.
Carlsson A, Lindqvist M (1973). Effect of ethanol on the hydroxylation of tyrosine and tryptophan in rat brain in vivo. J Pharm Pharmacol 25: 437–440.
Carr DB, Sesack SR (2000). Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci 15: 3864–3873.
Carroll KM, Fenton LR, Ball SA, Nich C, Frankforter TL, Shi J et al (2004). Efficacy of disulfiram and cognitive behavior therapy in cocaine-dependent outpatients: a randomized placebo-controlled trial. Arch Gen Psychiatry 61: 264–272.
Carroll KM, Nich C, Ball SA, McCance E, Frankforter TL, Rounsaville BJ (2000). One-year follow-up of disulfiram and psychotherapy for cocaine-alcohol users: sustained effects of treatment. Addiction 95: 1335–1349.
Carroll KM, Nich C, Ball SA, McCance E, Rounsavile BJ (1998). Treatment of cocaine and alcohol dependence with psychotherapy and disulfiram. Addiction 93: 713–727.
Carroll KM, Rounsaville BJ, Bryant KJ (1993). Alcoholism in treatment-seeking cocaine abusers: clinical and prognostic significance. J Stud Alcohol 54: 199–208.
Cervo L, Rossi C, Samanin R (1993). Clonidine-induced place preference is mediated by alpha 2-adrenoceptors outside the locus coeruleus. Eur J Pharmacol 238: 201–207.
Cohen C, Perrault G, Sanger DJ (1998). Preferential involvement of D3 versus D2 dopamine receptors in the effects of dopamine receptor ligands on oral ethanol self-administration in rats. Psychopharmacology (Berlin) 140: 478–485.
Cohen C, Perrault G, Sanger DJ (1999). Effects of D1 dopamine receptor agonists on oral ethanol self-administration in rats: comparison with their efficacy to produce grooming and hyperactivity. Psychopharmacology (Berlin) 142: 102–110.
Corcoran ME, Lewis J, Fibiger HC (1983). Forebrain noradrenaline and oral self-administration of ethanol by rats. Behav Brain Res 8: 1–21.
Corrodi H, Fuxe K, Hokfelt T (1966). The effect of ethanol on the activity of central catecholamine neurones in rat brain. J Pharm Pharmacol 18: 821–823.
Crow TJ (1970). Enhancement of coaine of intra-cranial self-stimulation in the rat. Life Sci 9: 375–381.
Crow TJ, Spear PJ, Arbuthnott GW (1972). Intracranial self-stimulation with electrodes in the region of the locus coeruleus. Brain Res 36: 275–287.
Cubells JF, Kranzler HR, McCance-Katz E, Anderson GM, Malison RT, Price LH et al (2000). A haplotype at the DBH locus, associated with low plasma dopamine beta-hydroxylase activity, also associates with cocaine-induced paranoia. Mol Psychiatry 5: 56–63.
D’Souza MS, Ikegami A, Olsen CM, Duvauchelle CL (2003). Chronic D1 agonist and ethanol coadministration facilitate ethanol-mediated behaviors. Pharmacol Biochem Behav 76: 335–342.
Dackis C, O’Brien C (2003). Glutamatergic agents for cocaine dependence. Ann NY Acad Sci 1003: 328–345.
Dackis CA, Kampman KM, Lynch KG, Pettinati HM, O’Brien CP (2005). A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology 30: 205–211.
Dackis CA, Lynch KG, Yu E, Samaha FF, Kampman KM, Cornish JW et al (2003). Modafinil and cocaine: a double-blind, placebo-controlled drug interaction study. Drug Alcohol Depend 70: 29–37.
Daoust M, Protais P, Ladure P (1990). Noradrenergic system: effect of DSP4 and FLA-57 on ethanol intake in ethanol preferring rats. Pharmacol Biochem Behav 36: 133–137.
Darracq L, Blanc G, Glowinski J, Tassin JP (1998). Importance of the noradrenaline–dopamine coupling in the locomotor activating effects of D-amphetamine. J Neurosci 18: 2729–2739.
Davis WM, Babbini M, Khalsa HJ (1972). Antagonism by alpha-methyltyrosine of morphine induced motility in non-tolerant and tolerant rats. Res Commun Chem Pathol Pharmacol 4: 267–279.
Davis WM, Smith SG (1977). Catecholaminergic mechanisms of reinforcement: direct assessment by drug-self-administration. Life Sci 20: 483–492.
Davis WM, Smith SG, Khalsa JH (1975). Noradrenergic role in the self-administration of morphine or amphetamine. Pharmacol Biochem Behav 3: 477–484.
Day NC, Wood SJ, Ince PG, Volsen SG, Smith W, Slater CR et al (1997). Differential localization of voltage-dependent calcium channel alpha1 subunits at the human and rat neuromuscular junction. J Neurosci 17: 6226–6235.
Delfs JM, Zhu Y, Druhan JP, Aston-Jones GS (1998). Origin of noradrenergic afferents to the shell subregion of the nucleus accumbens: anterograde and retrograde tract-tracing studies in the rat. Brain Res 806: 127–140.
Devoto P, Flore G, Pani L, Gessa GL (2001). Evidence for co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex. Mol Psychiatry 6: 657–664.
Devoto P, Flore G, Saba P, Fa M, Gessa GL (2005). Stimulation of the locus coeruleus elicits noradrenaline and dopamine release in the medial prefrontal and parietal cortex. J Neurochem 92: 368–374.
Di Chiara G, Tanda GL, Frau R, Carboni E (1992). Heterologous monoamine reuptake: lack of transmitter specificity of neuron-specific carriers. Neurochem Int 20 (Suppl): 231S–235S.
Dickinson SL, Gadie B, Tulloch IF (1988). Alpha 1- and alpha 2-adrenoreceptor antagonists differentially influence locomotor and stereotyped behaviour induced by D-amphetamine and apomorphine in the rat. Psychopharmacology (Berlin) 96: 521–527.
Domyancic AV, Morilak DA (1997). Distribution of alpha1A adrenergic receptor mRNA in the rat brain visualized by in situ hybridization. J Comp Neurol 386: 358–378.
Donaldson I, Dolphin A, Jenner P, Marsden CD, Pycock C (1976). The roles of noradrenaline and dopamine in contraversive circling behaviour seen after unilateral electrolytic lesions of the locus coeruleus. Eur J Pharmacol 39: 179–191.
Dresse A (1966). Importance of the noradrenergic mesencephalo-telencephalic system as an anatomic substrate of autostimulation behavior. Life Sci 5: 1003–1014.
Drouin C, Blanc G, Trovero F, Glowinski J, Tassin JP (2001). Cortical alpha 1-adrenergic regulation of acute and sensitized morphine locomotor effects. NeuroReport 12: 3483–3486.
Drouin C, Darracq L, Trovero F, Blanc G, Glowinski J, Cotecchia S et al (2002). Alpha1b-adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J Neurosci 22: 2873–2884.
Duteil J, Rambert FA, Pessonnier J, Hermant JF, Gombert R, Assous E (1990). Central alpha 1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur J Pharmacol 180: 49–58.
Dworkin SI, Guerin GF, Co C, Goeders NE, Smith JE (1988). Lack of an effect of 6-hydroxydopamine lesions of the nucleus accumbens on intravenous morphine self-administration. Pharmacol Biochem Behav 30: 1051–1057.
Dyr W, McBride WJ, Lumeng L, Li TK, Murphy JM (1993). Effects of D1 and D2 dopamine receptor agents on ethanol consumption in the high-alcohol-drinking (HAD) line of rats. Alcohol 10: 207–212.
Eidelberg E, Schwartz AS (1970). Possible mechanism of action of morphine on brain. Nature 225: 1152–1153.
El-Ghundi M, George SR, Drago J, Fletcher PJ, Fan T, Nguyen T et al (1998). Disruption of dopamine D1 receptor gene expression attenuates alcohol-seeking behavior. Eur J Pharmacol 353: 149–158.
Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J (2000). Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology 23: 138–150.
Estler CJ (1973). Effect of - and -adrenergic blocking agents and para-chlorophenylalanine on morphine- and caffeine-stimulated locomotor activity of mice. Psychopharmacologia 28: 261–268.
Ettenberg A, Pettit HO, Bloom FE, Koob GF (1982). Heroin and cocaine intravenous self-administration in rats: mediation by separate neural systems. Psychopharmacology (Berlin) 78: 204–209.
Ewing JA, Mueller RA, Rouse BA, Silver D (1977). Low levels of dopamine beta-hydroxylase and psychosis. Am J Psychiatry 134: 927–928.
Ewing JA, Rouse BA, Mueller RA, Silver D (1978). Can dopamine beta-hydroxylase levels predict adverse reactions to disulfiram? Alcohol Clin Exp Res 2: 93–94.
Ferraro L, Antonelli T, Tanganelli S, O’Connor WT, Perez de la Mora M, Mendez-Franco J et al (1999). The vigilance promoting drug modafinil increases extracellular glutamate levels in the medial preoptic area and the posterior hypothalamus of the conscious rat: prevention by local GABAA receptor blockade. Neuropsychopharmacology 20: 346–356.
Fibiger HC, Phillips AG (1974). Role of dopamine and norepinephrine in the chemistry of reward. J Psychiatr Res 11: 135–143.
Foote SL, Bloom FE, Aston-Jones G (1983). Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev 63: 844–914.
Fuller RK, Branchey L, Brightwell DR, Derman RM, Emrick CD, Iber FL et al (1986). Disulfiram treatment of alcoholism. A Veterans Administration cooperative study. JAMA 256: 1449–1455.
George TP, Chawarski MC, Pakes J, Carroll KM, Kosten TR, Schottenfeld RS (2000). Disulfiram versus placebo for cocaine dependence in buprenorphine-maintained subjects: a preliminary trial. Biol Psychiatry 47: 1080–1086.
Gerber GJ, Wise RA (1989). Pharmacological regulation of intravenous cocaine and heroin self-administration in rats: a variable dose paradigm. Pharmacol Biochem Behav 32: 527–531.
Gerrits MA, Ramsey NF, Wolterink G, van Ree JM (1994). Lack of evidence for an involvement of nucleus accumbens dopamine D1 receptors in the initiation of heroin self-administration in the rat. Psychopharmacology (Berlin) 114: 486–494.
Gerrits MA, Van Ree JM (1996). Effect of nucleus accumbens dopamine depletion on motivational aspects involved in initiation of cocaine and heroin self-administration in rats. Brain Res 713: 114–124.
Gibbs ME, Summers RJ (2002). Role of adrenoceptor subtypes in memory consolidation. Prog Neurobiol 67: 345–391.
Goldberg SR, Gonzalez FA (1976). Effects of propranolol on behavior maintained under fixed-ratio schedules of cocaine injection or food presentation in squirrel monkeys. J Pharmacol Exp Ther 198: 626–634.
Goldstein M, Anagnoste B, Lauber E, McKeregham MR (1964). Inhibition of dopamine-beta-hydroxylase by disulfiram. Life Sci 3: 763–767.
Gonon FG (1988). Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience 24: 19–28.
Gonzales RA, Weiss F (1998). Suppression of ethanol-reinforced behavior by naltrexone is associated with attenuation of the ethanol-induced increase in dialysate dopamine levels in the nucleus accumbens. J Neurosci 18: 10663–10671.
Goodwin FL, Koechling UM, Smith BR, Amit Z (1996). Lack of effect of dopamine D2 blockade on ethanol intake in selected and unselected strains of rats. Alcohol 13: 273–279.
Green AL (1964). The inhibition of dopamine-B-oxidase by chelating agents. Biochem Biophys Acta 81: 394–397.
Grenhoff J, Nisell M, Ferre S, Aston-Jones G, Svensson TH (1993). Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J Neural Transm Gen Sect 93: 11–25.
Grenhoff J, North RA, Johnson SW (1995). Alpha 1-adrenergic effects on dopamine neurons recorded intracellularly in the rat midbrain slice. Eur J Neurosci 7: 1707–1713.
Grenhoff J, Svensson TH (1989). Clonidine modulates dopamine cell firing in rat ventral tegmental area. Eur J Pharmacol 165: 11–18.
Grenhoff J, Svensson TH (1993). Prazosin modulates the firing pattern of dopamine neurons in rat ventral tegmental area. Eur J Pharmacol 233: 79–84.
Grosz HJ (1972). Successful treatment of a heroin addict with propranolol: implications for opiate addiction, treatment and research. J Indiana State Med Assoc 65: 505–509.
Grupp LA, Sneddon B, Solway E, Perlanski E, Stewart RB (1989). The beta adrenergic agonist isoproterenol suppress voluntary alcohol intake in rats. Pharmacol Biochem Behav 33: 493–495.
Hameedi FA, Rosen MI, McCance-Katz EF, McMahon TJ, Price LH, Jatlow PI et al (1995). Behavioral, physiological, and pharmacological interaction of cocaine and disulfiram in humans. Biol Psychiatry 37: 560–563.
Hand TH, Stinus L, Le Moal M (1989). Differential mechanisms in the acquisition and expression of heroin-induced place preference. Psychopharmacology (Berlin) 98: 61–67.
Harris GC, Hedaya MA, Pan WJ, Kalivas P (1996). beta-adrenergic antagonism alters the behavioral and neurochemical responses to cocaine. Neuropsychopharmacology 14: 195–204.
Harro J, Merikula A, Lepiku M, Modiri AR, Rinken A, Oreland L (2000). Lesioning of locus coeruleus projections by DSP-4 neurotoxin treatment: effect on amphetamine-induced hyperlocomotion and dopamine D2 receptor binding in rats. Pharmacol Toxicol 86: 197–202.
Heath RG, Nesselhof Jr W, Bishop MP, Byers LW (1965). Behavioral and metabolic changes associated with administration of tetraethylthiuram disulfide (Antabuse). Dis Nerv Syst 26: 99–105.
Hermant JF, Rambert FA, Duteil J (1991). Awakening properties of modafinil: effect on nocturnal activity in monkeys (Macaca mulatta) after acute and repeated administration. Psychopharmacology (Berlin) 103: 28–32.
Higgins ST, Budney AJ, Bickel WK, Hughes JR, Foerg F (1993). Disulfiram therapy in patients abusing cocaine and alcohol. Am J Psychiatry 150: 675–676.
Hnasko TS, Sotak BN, Palmiter RD (2005). Morphine reward in dopamine-deficient mice. Nature 438: 854–857.
Hoeldtke RD, Stetson PL (1980). An in vivo tritium release assay of human dopamine beta-hydroxylase. J Clin Endocrinol Metab 51: 810–815.
Howell LL, Byrd LD (1991). Characterization of the effects of cocaine and GBR 12909, a dopamine uptake inhibitor, on behavior in the squirrel monkey. J Pharmacol Exp Ther 258: 178–185.
Hubbell CL, Marglin SH, Spitalnic SJ, Abelson ML, Wild KD, Reid LD (1991). Opioidergic, serotonergic, and dopaminergic manipulations and rats’ intake of a sweetened alcoholic beverage. Alcohol 8: 355–367.
Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C (2003). Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem 84: 698–704.
Hunt WA, Majchrowicz E (1974). Alterations in the turnover of brain norepinephrine and dopamine in alcohol-dependent rats. J Neurochem 23: 549–552.
Ikemoto S, Glazier BS, Murphy JM, McBride WJ (1997). Role of dopamine D1 and D2 receptors in the nucleus accumbens in mediating reward. J Neurosci 17: 8580–8587.
Jacob RG, Holmstrand J, Eriksson JH, Anggard E (1975). Propranolol in the treatment of opiate dependence—a controlled study. Psychopharmacologia 41: 71–73.
Jasmin L, Tien D, Weinshenker D, Palmiter RD, Green PG, Janni G et al (2002). The NK1 receptor mediates both the hyperalgesia and the resistance to morphine in mice lacking noradrenaline. Proc Natl Acad Sci USA 99: 1029–1034.
Jones BE, Halaris AE, McIlhany M, Moore RY (1977). Ascending projections of the locus coeruleus in the rat. I. Axonal transport in central noradrenaline neurons. Brain Res 127: 1–21.
Jones CR, Hamilton CA, Whyte KF, Elliott HL, Reid JL (1985). Acute and chronic regulation of alpha 2-adrenoceptor number and function in man. Clin Sci (London) 68 (Suppl 10): 129s–132s.
Karamanakos PN, Pappas P, Stephanou P, Marselos M (2001). Differentiation of disulfiram effects on central catecholamines and hepatic ethanol metabolism. Pharmacol Toxicol 88: 106–110.
Karoum F, Wyatt RJ, Majchrowicz E (1976). Brain concentrations of biogenic amine metabolites in acutely treated and ethanol-dependent rats. Br J Pharmacol 56: 403–411.
Kiianmaa K (1980). Alcohol intake and ethanol intoxication in the rate: effect of a 6-OHDA-induced lesion of the ascending noradrenaline pathways. Eur J Pharmacol 64: 9–19.
Kiianmaa K, Andersson K, Fuxe K (1979). On the role of ascending dopamine systems in the control of voluntary ethanol intake and ethanol intoxication. Pharmacol Biochem Behav 10: 603–608.
Kohnke MD, Kolb W, Kohnke AM, Lutz U, Schick S, Batra A (2006). DBH(*)444G/A polymorphism of the dopamine-beta-hydroxylase gene is associated with alcoholism but not with severe alcohol withdrawal symptoms. J Neural Transm 113: 869–876.
Kohnke MD, Zabetian CP, Anderson GM, Kolb W, Gaertner I, Buchkremer G et al (2002). A genotype-controlled analysis of plasma dopamine beta-hydroxylase in healthy and alcoholic subjects: evidence for alcohol-related differences in noradrenergic function. Biol Psychiatry 52: 1151–1158.
Koistinen M, Tuomainen P, Hyytia P, Kiianmaa K (2001). Naltrexone suppresses ethanol intake in 6-hydroxydopamine-treated rats. Alcohol Clin Exp Res 25: 1605–1612.
Koob GF, Rassnick S, Heinrichs S, Weiss F (1994). Alcohol, the reward system and dependence. Alcohol Clin Exp Res 71: 103–114.
Koob GF, Roberts AJ, Schulteis G, Parsons LH, Heyser CJ, Hyytia P et al (1998). Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res 22: 3–9.
Kosten T, Sofuoglu M, Poling J, Gonsai K, Oliveto A (2005). Desipramine treatment for cocaine dependence in buprenorphine- or methadone-treated patients: baseline urine results as predictor of response. Am J Addict 14: 8–17.
Lategan AJ, Marien MR, Colpaert FC (1990). Effects of locus coeruleus lesions on the release of endogenous dopamine in the rat nucleus accumbens and caudate nucleus as determined by intracerebral microdialysis. Brain Res 523: 134–138.
Lategan AJ, Marien MR, Colpaert FC (1992). Suppression of nigrostriatal and mesolimbic dopamine release in vivo following noradrenaline depletion by DSP-4: a microdialysis study. Life Sci 50: 995–999.
Laviolette SR, Nader K, van der Kooy D (2002). Motivational state determines the functional role of the mesolimbic dopamine system in the mediation of opiate reward processes. Behav Brain Res 129: 17–29.
Le AD, Harding S, Juzytsch W, Funk D, Shaham Y (2005). Role of alpha-2 adrenoceptors in stress-induced reinstatement of alcohol seeking and alcohol self-administration in rats. Psychopharmacology (Berlin) 179: 366–373.
Lee B, Tiefenbacher S, Platt DM, Spealman RD (2004). Pharmacological blockade of alpha2-adrenoceptors induces reinstatement of cocaine-seeking behavior in squirrel monkeys. Neuropsychopharmacology 29: 686–693.
Leri F, Flores J, Rodaros D, Stewart J (2002). Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J Neurosci 22: 5713–5718.
Letchworth SR, Nader MA, Smith HR, Friedman DP, Porrino LJ (2001). Progression of changes in dopamine transporter binding site density as a result of cocaine self-administration in rhesus monkeys. J Neurosci 21: 2799–2807.
Levine ES, Litto WJ, Jacobs BL (1990). Activity of cat locus coeruleus noradrenergic neurons during the defense reaction. Brain Res 531: 189–195.
Lindvall O, Björklund A (1974). The organization of the ascending catecholamine neuron systems in the rat brain as revealed by the glyoxylic acid fluorescence method. Acta Physiol Scand Suppl 412: 1–48.
Liprando LA, Miner LH, Blakely RD, Lewis DA, Sesack SR (2004). Ultrastructural interactions between terminals expressing the norepinephrine transporter and dopamine neurons in the rat and monkey ventral tegmental area. Synapse 52: 233–244.
Lu L, Su WJ, Yue W, Ge X, Su F, Pei G et al (2001). Attenuation of morphine dependence and withdrawal in rats by venlafaxine, a serotonin and noradrenaline reuptake inhibitor. Life Sci 69: 37–46.
Lyness WH, Smith FL (1992). Influence of dopaminergic and serotonergic neurons on intravenous ethanol self-administration in the rat. Pharmacol Biochem Behav 42: 187–192.
Macey DJ, Smith HR, Nader MA, Porrino LJ (2003). Chronic cocaine self-administration upregulates the norepinephrine transporter and alters functional activity in the bed nucleus of the stria terminalis of the rhesus monkey. J Neurosci 23: 12–16.
Madras BK, Xie Z, Lin Z, Jassen AJ, Panas H, Lynch L et al (2006). Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther 319: 561–569.
Major LF, Lerner P, Ballenger JC, Brown GL, Goodwin FK, Lovenberg W (1979). Dopamine-beta-hydroxylase in the cerebrospinal fluid: relationship to disulfiram-induced psychosis. Biol Psychiatry 14: 337–344.
Maldonado R (1997). Participation of noradrenergic pathways in the expression of opiate withdrawal: biochemical and pharmacological evidence. Neurosci Biobehav Rev 21: 91–104.
Marek GJ, Aghajanian GK (1999). 5-HT2A receptor or alpha1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur J Pharmacol 367: 197–206.
Mason ST, Corcoran ME, Fibiger HC (1979). Noradrenaline and ethanol intake in the rat. Neurosci Lett 12: 137–142.
Matchett JA, Erickson CK (1977). Alteration of ethanol-induced changes in locomotor activity by adrenergic blockers in mice. Psychopharmacology (Berlin) 52: 201–206.
Mays DC, Ortiz-Bermudez P, Lam JP, Tong IH, Fauq AH, Lipsky JJ (1998). Inhibition of recombinant human mitochondrial aldehyde dehydrogenase by two intermediate metabolites of disulfiram. Biochem Pharmacol 55: 1099–1103.
McBride WJ, Murphy JM, Ikemoto S (1999). Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 101: 129–152.
McCance-Katz EF, Kosten TR, Jatlow P (1998a). Chronic disulfiram treatment effects on intranasal cocaine administration: initial results. Biol Psychiatry 43: 540–543.
McCance-Katz EF, Kosten TR, Jatlow P (1998b). Disulfiram effects on acute cocaine administration. Drug Alcohol Depend 52: 27–39.
McDowell D, Nunes EV, Seracini AM, Rothenberg J, Vosburg SK, Ma GJ et al (2005). Desipramine treatment of cocaine-dependent patients with depression: a placebo-controlled trial. Drug Alcohol Depend 80: 209–221.
Melchior CL, Myers RD (1976). Genetic differences in ethanol drinking of the rat following injection of 6-OHDA, 5,6-DHT or 5,7-DHT into the cerebral ventricles. Pharmacol Biochem Behav 5: 63–72.
Melendez RI, Rodd-Henricks ZA, Engleman EA, Li TK, McBride WJ, Murphy JM (2002). Microdialysis of dopamine in the nucleus accumbens of alcohol-preferring (P) rats during anticipation and operant self-administration of ethanol. Alcohol Clin Exp Res 26: 318–325.
Miner LL, Drago J, Chamberlain PM, Donovan D, Uhl GR (1995). Retained cocaine conditioned place preference in D1 receptor deficient mice. NeuroReport 6: 2314–2316.
Mohammed AK, Danysz W, Ogren SO, Archer T (1986). Central noradrenaline depletion attenuates amphetamine-induced locomotor behavior. Neurosci Lett 64: 139–144.
Moore RY, Bloom FE (1979). Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci 2: 113–168.
Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT (2002). Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci 22: 389–395.
Morrison JH, Molliver ME, Grzanna R, Coyle JT (1981). The intra-cortical trajectory of the coeruleo-cortical projection in the rat: a tangentially organized cortical afferent. Neuroscience 6: 139–158.
Mundorf ML, Joseph JD, Austin CM, Caron MG, Wightman RM (2001). Catecholamine release and uptake in the mouse prefrontal cortex. J Neurochem 79: 130–142.
Murchison CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA (2004). A distinct role for norepinephrine in memory retrieval. Cell 117: 131–143.
Musacchio JM, Goldstein M, Anagnoste B, Poch G, Kopin IJ (1966). Inhibition of dopamine-beta-hydroxylase by disulfiram in vivo. J Pharmacol Exp Ther 152: 56–61.
Nader MA, Daunais JB, Moore T, Nader SH, Moore RJ, Smith HR et al (2002). Effects of cocaine self-administration on striatal dopamine systems in rhesus monkeys: initial and chronic exposure. Neuropsychopharmacology 27: 35–46.
Nestler EJ, Alreja M, Aghajanian GK (1994). Molecular and cellular mechanisms of opiate action: studies in the rat locus coeruleus. Brain Res Bull 35: 521–528.
Nestler EJ, Alreja M, Aghajanian GK (1999). Molecular control of locus coeruleus neurotransmission. Biol Psychiatry 46: 1131–1139.
Ng GY, George SR (1994). Dopamine receptor agonist reduces ethanol self-administration in the ethanol-preferring C57BL/6J inbred mouse. Eur J Pharmacol 269: 365–374.
Nicholas AP, Pieribone V, Hokfelt T (1993). Distributions of mRNAs for alpha-2 adrenergic receptor subtypes in rat brain: an in situ hybridization study. J Comp Neurol 328: 575–594.
Nicola SM, Malenka RC (1998). Modulation of synaptic transmission by dopamine and norepinephrine in ventral but not dorsal striatum. J Neurophysiol 79: 1768–1776.
Nurmi M, Sinclair JD, Kiianmaa K (1998). Dopamine release during ethanol drinking in AA rats. Alcohol Clin Exp Res 22: 1628–1633.
Olds J, Milner P (1954). Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47: 419–427.
Olive MF, Mehmert KK, Messing RO, Hodge CW (2000). Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKCepsilon-deficient mice. Eur J Neurosci 12: 4131–4140.
Olson VG, Heusner CL, Bland RJ, During MJ, Weinshenker D, Palmiter RD (2006). Role of noradrenergic signaling by the nucleus tractus solitarius in mediating opiate reward. Science 311: 1017–1020.
Ordway GA, Jaconetta SM, Halaris AE (1993). Characterization of subtypes of alpha-2 adrenoceptors in the human brain. J Pharmacol Exp Ther 264: 967–976.
Palacios JM, Hoyer D, Cortes R (1987). alpha 1-Adrenoceptors in the mammalian brain: similar pharmacology but different distribution in rodents and primates. Brain Res 419: 65–75.
Paladini CA, Fiorillo CD, Morikawa H, Williams JT (2001). Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nat Neurosci 4: 275–281.
Paladini CA, Williams JT (2004). Noradrenergic inhibition of midbrain dopamine neurons. J Neurosci 24: 4568–4575.
Paradisi R, Grossi G, Pintore A, Venturoli S, Porcu E, Capelli M et al (1991). Evidence for a pathological reduction in brain dopamine metabolism in idiopathic hyperprolactinemia. Acta Endocrinol (Copenhagen) 125: 246–252.
Petrakis IL, Carroll KM, Nich C, Gordon LT, McCance-Katz EF, Frankforter T et al (2000). Disulfiram treatment for cocaine dependence in methadone-maintained opioid addicts. Addiction 95: 219–228.
Pettit HO, Ettenberg A, Bloom FE, Koob GF (1984). Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self-administration in rats. Psychopharmacology (Berlin) 84: 167–173.
Phillips TJ, Brown KJ, Burkhart-Kasch S, Wenger CD, Kelly MA, Rubinstein M et al (1998). Alcohol preference and sensitivity are markedly reduced in mice lacking dopamine D2 receptors. Nat Neurosci 1: 610–615.
Pierce RC, Kumaresan V (2006). The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev 30: 215–238.
Pieribone VA, Nicholas AP, Dagerlind A, Hokfelt T (1994). Distribution of alpha 1 adrenoceptors in rat brain revealed by in situ hybridization experiments utilizing subtype-specific probes. J Neurosci 14: 4252–4268.
Pijnenburg AJ, Honig WM, Van Rossum JM (1975). Inhibition of D-amphetamine-induced locomotor activity by injection of haloperidol into the nucleus accumbens of the rat. Psychopharmacologia 41: 87–95.
Pohorecky LA, Brick J (1988). Pharmacology of ethanol. Pharmacol Ther 36: 335–427.
Pohorecky LA, Jaffe LS (1975). Noradrenergic involvement in the acute effects of ethanol. Res Commun Chem Pathol Pharmacol 12: 433–447.
Porrino LJ, Lyons D, Miller MD, Smith HR, Friedman DP, Daunais JB et al (2002). Metabolic mapping of the effects of cocaine during the initial phases of self-administration in the nonhuman primate. J Neurosci 22: 7687–7694.
Poschel BP, Ninteman FW (1963). Norepinephrine: a possible excitatory neurohormone of the reward system. Life Sci 10: 782–788.
Pozuelo J, Kerr FW (1972). Suppression of craving and other signs of dependence in morphine-addicted monkeys by administration of alpha-methyl-para-tyrosine. Mayo Clin Proc 47: 621–628.
Rainbow TC, Biegon A (1983). Quantitative autoradiography of [3H]prazosin binding sites in rat forebrain. Neurosci Lett 40: 221–226.
Raiteri M, Del Carmine R, Bertollini A, Levi G (1977). Effect of sympathomimetic amines on the synaptosomal transport of noradrenaline, dopamine and 5-hydroxytryptamine. Eur J Pharmacol 41: 133–143.
Rassnick S, Pulvirenti L, Koob GF (1993a). SDZ-205,152, a novel dopamine receptor agonist, reduces oral ethanol self-administration in rats. Alcohol 10: 127–132.
Rassnick S, Stinus L, Koob GF (1993b). The effects of 6-hydroxydopamine lesions of the nucleus accumbens and the mesolimbic dopamine system on oral self-administration of ethanol in the rat. Brain Res 623: 16–24.
Regier DA, Farmer ME, Rae DS, Locke BZ, Keith SJ, Judd LL et al (1990). Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA 264: 2511–2518.
Resnick RB, Kestenbaum RS, Schwartz LK, Smith A (1976). Evaluation of propranolol in opiate dependence. Arch Gen Psychiatry 33: 993–997.
Ressler KJ, Nemeroff CB (1999). Role of norepinephrine in the pathophysiology and treatment of mood disorders. Biol Psychiatry 46: 1219–1233.
Richardson JS, Novakovski DM (1978). Brain monoamines and free choice ethanol consumption in rats. Drug Alcohol Depend 3: 253–264.
Risinger FO, Freeman PA, Rubinstein M, Low MJ, Grandy DK (2000). Lack of operant ethanol self-administration in dopamine D2 receptor knockout mice. Psychopharmacology (Berlin) 152: 343–350.
Ritter S, Stein L (1973). Self-stimulation of noradrenergic cell group (A6) in locus coeruleus of rats. J Comp Physiol Psychol 85: 443–452.
Ritter S, Stein L (1974). Self-stimulation in the mesencephalic trajectory of the ventral noradrenergic bundle. Brain Res 81: 145–157.
Roberts DC, Corcoran ME, Fibiger HC (1977). On the role of ascending catecholaminergic systems in intravenous self-administration of cocaine. Pharmacol Biochem Behav 6: 615–620.
Roberts DC, Mason ST, Fibiger HC (1978). 6-OHDA lesion to the dorsal noradrenergic bundle alters morphine-induced locomotor activity and catalepsy. Eur J Pharmacol 52: 209–214.
Robinson TE, Berridge KC (2000). The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction 95 (Suppl 2): S91–S117.
Rocha BA (2003). Stimulant and reinforcing effects of cocaine in monoamine transporter knockout mice. Eur J Pharmacol 479: 107–115.
Rogers WK, Benowitz NL, Wilson KM, Abbott JA (1979). Effect of disulfiram on adrenergic function. Clin Pharmacol Ther 25: 469–477.
Rosen GF, Lobo RA (1987). Further evidence against dopamine deficiency as the cause of inappropriate gonadotropin secretion in patients with polycystic ovary syndrome. J Clin Endocrinol Metab 65: 891–895.
Russell VA, Lamm MC, Allin R, de Villiers AS, Searson A, Taljaard JJ (1989). Effect of selective noradrenergic denervation on noradrenaline content and [3H]dopamine release in rat nucleus accumbens slices. Neurochem Res 14: 169–172.
Sahraei H, Ghazzaghi H, Zarrindast MR, Ghoshooni H, Sepehri H, Haeri-Rohan A (2004). The role of alpha-adrenoceptor mechanism(s) in morphine-induced conditioned place preference in female mice. Pharmacol Biochem Behav 78: 135–141.
Salomon L, Lanteri C, Glowinski J, Tassin JP (2006). Behavioral sensitization to amphetamine results from an uncoupling between noradrenergic and serotonergic neurons. Proc Natl Acad Sci USA 103: 7476–7481.
Scatton B, Dennis T, Curet O (1984). Increase in dopamine and DOPAC levels in noradrenergic terminals after electrical stimulation of the ascending noradrenergic pathways. Brain Res 298: 193–196.
Schank JR, Ventura R, Puglisi-Allegra S, Alcaro A, Cole CD, Liles LC et al (2006). Dopamine beta-hydroxylase knockout mice have alterations in dopamine signaling and are hypersensitive to cocaine. Neuropsychopharmacology 31: 2221–2230.
Schwartz AS, Marchok PL (1974). Depression of morphine-seeking behaviour by dopamine inhibition. Nature 248: 257–258.
Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK et al (2005). Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis. Proc Natl Acad Sci USA 102: 3513–3518.
Shaham Y, Highfield D, Delfs J, Leung S, Stewart J (2000). Clonidine blocks stress-induced reinstatement of heroin seeking in rats: an effect independent of locus coeruleus noradrenergic neurons. Eur J Neurosci 12: 292–302.
Shaham Y, Shalev U, Lu L, De Wit H, Stewart J (2003). The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berlin) 168: 3–20.
Shearman GT, Hynes M, Lal H (1981). Self-administration of clonidine by the rat. Prog Clin Biol Res 71: 259–276.
Shi WX, Pun CL, Zhang XX, Jones MD, Bunney BS (2000). Dual effects of D-amphetamine on dopamine neurons mediated by dopamine and nondopamine receptors. J Neurosci 20: 3504–3511.
Silvestre JS, O’Neill MF, Fernandez AG, Palacios JM (1996). Effects of a range of dopamine receptor agonists and antagonists on ethanol intake in the rat. Eur J Pharmacol 318: 257–265.
Simon H, Le Moal M, Cardo B (1975). Self-stimulation in the dorsal pontine tegmentum in the rat. Behav Biol 13: 339–347.
Simon H, Le Moal M, Stinus L, Calas A (1979). Anatomical relationships between the ventral mesencephalic tegmentum—a 10 region and the locus coeruleus as demonstrated by anterograde and retrograde tracing techniques. J Neural Transm 44: 77–86.
Singer G, Wallace M (1984). Effects of 6-OHDA lesions in the nucleus accumbens on the acquisition of self injection of heroin under schedule and non schedule conditions in rats. Pharmacol Biochem Behav 20: 807–809.
Skjoldager P, Winger G, Woods JH (1993). Effects of GBR 12909 and cocaine on cocaine-maintained behavior in rhesus monkeys. Drug Alcohol Depend 33: 31–39.
Smith JE, Guerin GF, Co C, Barr TS, Lane JD (1985). Effects of 6-OHDA lesions of the central medial nucleus accumbens on rat intravenous morphine self-administration. Pharmacol Biochem Behav 23: 843–849.
Snoddy AM, Tessel RE (1985). Prazosin: effect on psychomotor-stimulant cues and locomotor activity in mice. Eur J Pharmacol 116: 221–228.
Sofuoglu M, Kosten TR (2006). Emerging pharmacological strategies in the fight against cocaine addiction. Expert Opin Emerg Drugs 11: 91–98.
Sommermeyer H, Frielingsdorf J, Knorr A (1995). Effects of prazosin on the dopaminergic neurotransmission in rat brain. Eur J Pharmacol 276: 267–270.
Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R et al (1998). Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci USA 95: 7699–7704.
Spyraki C, Fibiger HC, Phillips AG (1982a). Cocaine-induced place preference conditioning: lack of effects of neuroleptics and 6-hydroxydopamine lesions. Brain Res 253: 195–203.
Spyraki C, Fibiger HC, Phillips AG (1982b). Dopaminergic substrates of amphetamine-induced place preference conditioning. Brain Res 253: 185–193.
Spyraki C, Fibiger HC, Phillips AG (1983). Attenuation of heroin reward in rats by disruption of the mesolimbic dopamine system. Psychopharmacology (Berlin) 79: 278–283.
Stein L (1964). Self-stimulation of the brain and the central stimulant action of amphetamine. Fed Proc 23: 836–850.
Stein L, Wise CD (1969). Release of norepinephrine from hypothalamus and amygdala by rewarding medial forebrain bundle stimulation and amphetamine. J Comp Physiol Psychol 67: 189–198.
Stone EA, Cotecchia S, Lin Y, Quartermain D (2002). Role of brain alpha 1B-adrenoceptors in modafinil-induced behavioral activity. Synapse 46: 269–270.
Stone EA, Lin Y, Ahsan R, Quartermain D (2004). Gross mapping of alpha1-adrenoceptors that regulate behavioral activation in the mouse brain. Behav Brain Res 152: 167–175.
Svensson L, Ahlenius S (1982). Functional importance of nucleus accumbens noradrenaline in the rat. Acta Pharmacol Toxicol (Copenhagen) 50: 22–24.
Swanson LW, Hartman BK (1975). The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol 163: 467–505.
Szerman N, Peris L, Mesias B, Colis P, Rosa J, Prieto A (2005). Reboxetine for the treatment of patients with cocaine dependence disorder. Hum Psychopharmacol 20: 189–192.
Takahashi S, Gjessing LR (1972). Studies of periodic catatonia. IV. Longitudinal study of catecholamine metabolism, with and without drugs. J Psychiatr Res 9: 293–314.
Tassin JP, Lavielle S, Herve D, Blanc G, Thierry AM, Alvarez C et al (1979). Collateral sprouting and reduced activity of the rat mesocortical dopaminergic neurons after selective destruction of the ascending noradrenergic bundles. Neuroscience 4: 1569–1582.
Tella SR (1995). Effects of monoamine reuptake inhibitors on cocaine self-administration in rats. Pharmacol Biochem Behav 51: 687–692.
Thomas SA, Marck BT, Palmiter RD, Matsumoto AM (1998). Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem 70: 2468–2476.
Thomas SA, Matsumoto AM, Palmiter RD (1995). Noradrenaline is essential for mouse fetal development. Nature 374: 643–646.
Tong J, Hornykiewicz O, Kish SJ (2006). Identification of a noradrenaline-rich subdivision of the human nucleus accumbens. J Neurochem 96: 349–354.
Tzschentke TM (1998). Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56: 613–672.
Tzschentke TM, Schmidt WJ (1998). Discrete quinolinic acid lesions of the rat prelimbic medial prefrontal cortex affect cocaine- and MK-801-, but not morphine- and amphetamine-induced reward and psychomotor activation as measured with the place preference conditioning paradigm. Behav Brain Res 97: 115–127.
Udenfriend S (1966). Tyrosine hydroxylase. Pharmacol Rev 18: 43–51.
Uhl GR, Hall FS, Sora I (2002). Cocaine, reward, movement and monoamine transporters. Mol Psychiatry 7: 21–26.
Uhlen S, Lindblom J, Tiger G, Wikberg JE (1997). Quantification of alpha2A and alpha2C adrenoceptors in the rat striatum and in different regions of the spinal cord. Acta Physiol Scand 160: 407–412.
Van Bockstaele EJ, Menko AS, Drolet G (2001). Neuroadaptive responses in brainstem noradrenergic nuclei following chronic morphine exposure. Mol Neurobiol 23: 155–171.
Van Ree JM, Ramsey N (1987). The dopamine hypothesis of opiate reward challenged. Eur J Pharmacol 134: 239–243.
Vanderschuren LJ, Wardeh G, De Vries TJ, Mulder AH, Schoffelmeer AN (1999). Opposing role of dopamine D1 and D2 receptors in modulation of rat nucleus accumbens noradrenaline release. J Neurosci 19: 4123–4131.
Ventura R, Alcaro A, Puglisi-Allegra S (2005). Prefrontal cortical norepinephrine release is critical for morphine-induced reward, reinstatement and dopamine release in the nucleus accumbens. Cereb Cortex 15: 1877–1886.
Ventura R, Cabib S, Alcaro A, Orsini C, Puglisi-Allegra S (2003). Norepinephrine in the prefrontal cortex is critical for amphetamine-induced reward and mesoaccumbens dopamine release. J Neurosci 23: 1879–1885.
Verbanck P, Seutin V, Dresse A, Scuvee J, Massotte L, Giesbers I et al (1990). Electrophysiological effects of ethanol on monoaminergic neurons: an in vivo and in vitro study. Alcohol Clin Exp Res 14: 728–735.
Villégier AS, Drouin C, Bizot JC, Marien M, Glowinski J, Colpaert F et al (2003). Stimulation of postsynaptic alpha1b- and alpha2-adrenergic receptors amplifies dopamine-mediated locomotor activity in both rats and mice. Synapse 50: 277–284.
Wang X, Cen X, Lu L (2001). Noradrenaline in the bed nucleus of the stria terminalis is critical for stress-induced reactivation of morphine-conditioned place preference in rats. Eur J Pharmacol 432: 153–161.
Wee S, Wang Z, He R, Zhou J, Kozikowski AP, Woolverton WL (2006). Role of the increased noradrenergic neurotransmission in drug self-administration. Drug Alcohol Depend 82: 151–157.
Weinshenker D, Miller NS, Blizinsky K, Laughlin ML, Palmiter RD (2002a). Mice with chronic norepinephrine deficiency resemble amphetamine-sensitized animals. Proc Natl Acad Sci USA 99: 13873–13877.
Weinshenker D, Rust NC, Miller NS, Palmiter RD (2000). Ethanol-associated behaviors of mice lacking norepinephrine. J Neurosci 20: 3157–3164.
Weinshenker D, Szot P, Miller NS, Palmiter RD (2001). Alpha(1) and beta(2) adrenoreceptor agonists inhibit pentylenetetrazole-induced seizures in mice lacking norepinephrine. J Pharmacol Exp Ther 298: 1042–1048.
Weinshenker D, White SS, Javors MA, Palmiter RD, Szot P (2002b). Regulation of norepinephrine transporter abundance by catecholamines and desipramine in vivo. Brain Res 946: 239–246.
Weiss F, Lorang MT, Bloom FE, Koob GF (1993). Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther 267: 250–258.
Weiss F, Mitchiner M, Bloom FE, Koob GF (1990). Free-choice responding for ethanol versus water in alcohol preferring (P) and unselected Wistar rats is differentially modified by naloxone, bromocriptine, and methysergide. Psychopharmacology (Berlin) 101: 178–186.
Weiss F, Parsons LH, Schulteis G, Hyytia P, Lorang MT, Bloom FE et al (1996). Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. J Neurosci 16: 3474–3485.
Wellman P, Ho D, Cepeda-Benito A, Bellinger L, Nation J (2002). Cocaine-induced hypophagia and hyperlocomotion in rats are attenuated by prazosin. Eur J Pharmacol 455: 117–126.
Wise CD, Berger BD, Stein L (1973). Evidence of α-noradrenergic reward receptors and serotonergic punishment receptors in the rat brain. Biol Psychiatry 6: 3–21.
Wise CD, Stein L (1969). Facilitation of brain self-stimulation by central administration of norepinephrine. Science 163: 299–301.
Wise RA (1978). Catecholamine theories of reward: a critical review. Brain Res 152: 215–247.
Wise RA (1980). Action of drugs of abuse on brain reward systems. Pharmacol Biochem Behav 13 (Suppl 1): 213–223.
Woolverton WL (1987). Evaluation of the role of norepinephrine in the reinforcing effects of psychomotor stimulants in rhesus monkeys. Pharmacol Biochem Behav 26: 835–839.
Woolverton WL, Wessinger WD, Balster RL (1982). Reinforcing properties of clonidine in rhesus monkeys. Psychopharmacology (Berlin) 77: 17–23.
Xu F, Gainetdinov RR, Wetsel WC, Jones SR, Bohn LM, Miller GW et al (2000). Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nat Neurosci 3: 465–471.
Yokel RA, Wise RA (1975). Increased lever pressing for amphetamine after pimozide in rats: implications for a dopamine theory of reward. Science 187: 547–549.
Yokel RA, Wise RA (1976). Attenuation of intravenous amphetamine reinforcement by central dopamine blockade in rats. Psychopharmacology (Berlin) 48: 311–318.
Zabetian CP, Anderson GM, Buxbaum SG, Elston RC, Ichinose H, Nagatsu T et al (2001). A quantitative-trait analysis of human plasma-dopamine beta-hydroxylase activity: evidence for a major functional polymorphism at the DBH locus. Am J Hum Genet 68: 515–522.
Zarrindast MR, Bahreini T, Adl M (2002). Effect of imipramine on the expression and acquisition of morphine-induced conditioned place preference in mice. Pharmacol Biochem Behav 73: 941–949.
Zhang W, Klimek V, Farley JT, Zhu MY, Ordway GA (1999). alpha2C adrenoceptors inhibit adenylyl cyclase in mouse striatum: potential activation by dopamine. J Pharmacol Exp Ther 289: 1286–1292.
Zhang W, Ordway GA (2003). The alpha2C-adrenoceptor modulates GABA release in mouse striatum. Brain Res Mol Brain Res 112: 24–32.
Zhang WP, Ouyang M, Thomas SA (2004). Potency of catecholamines and other L-tyrosine derivatives at the cloned mouse adrenergic receptors. Neuropharmacology 47: 438–449.
Zhang XY, Kosten TA (2005). Prazosin, an alpha-1 adrenergic antagonist, reduces cocaine-induced reinstatement of drug-seeking. Biol Psychiatry 57: 1202–1204.
Zhou Y, Bunney BS, Shi WX (2006). Differential effects of cocaine on firing rate and pattern of dopamine neurons: role of alpha1 receptors and comparison with L-dopa and apomorphine. J Pharmacol Exp Ther 317: 196–201.
Zito KA, Vickers G, Roberts DC (1985). Disruption of cocaine and heroin self-administration following kainic acid lesions of the nucleus accumbens. Pharmacol Biochem Behav 23: 1029–1036.
Zocchi A, Girlanda E, Varnier G, Sartori I, Zanetti L, Wildish GA et al (2003). Dopamine responsiveness to drugs of abuse: a shell–core investigation in the nucleus accumbens of the mouse. Synapse 50: 293–302.
Acknowledgements
We thank C Strauss, J Schank, and L Howell for critical reading of the manuscript and helpful suggestions. DW has received research funds from Cephalon Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Weinshenker, D., Schroeder, J. There and Back Again: A Tale of Norepinephrine and Drug Addiction. Neuropsychopharmacol 32, 1433–1451 (2007). https://doi.org/10.1038/sj.npp.1301263
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301263
Keywords
This article is cited by
-
Time estimation and arousal responses in dopa-responsive dystonia
Scientific Reports (2022)
-
Novel Pharmacological Agents for the Treatment of Cocaine Use Disorder
Current Behavioral Neuroscience Reports (2022)
-
Blunted Amphetamine-induced Reinforcing Behaviors and Transporter Downregulation in Knock-in Mice Carrying Alanine Mutations at Threonine-258 and Serine-259 of Norepinephrine Transporter
Journal of Molecular Neuroscience (2022)
-
Noradrenergic correlates of chronic cocaine craving: neuromelanin and functional brain imaging
Neuropsychopharmacology (2021)
-
Daidzein modulates cocaine-reinforcing effects and cue-induced cocaine reinstatement in CD-1 male mice
Psychopharmacology (2021)
