
Abstract
Noncompetitive N-methyl-D-aspartate (NMDA) blockers induce schizophrenic-like symptoms in humans, presumably by impairing glutamatergic transmission. Therefore, a compound potentiating this neurotransmission, by increasing extracellular levels of glycine (a requisite co-agonist of glutamate), could possess antipsychotic activity. Blocking the glycine transporter-1 (GlyT1) should, by increasing extracellular glycine levels, potentiate glutamatergic neurotransmission. SSR504734, a selective and reversible inhibitor of human, rat, and mouse GlyT1 (IC50=18, 15, and 38 nM, respectively), blocked reversibly the ex vivo uptake of glycine (mouse cortical homogenates: ID50: 5 mg/kg i.p.), rapidly and for a long duration. In vivo, it increased (minimal efficacious dose (MED): 3 mg/kg i.p.) extracellular levels of glycine in the rat prefrontal cortex (PFC). This resulted in an enhanced glutamatergic neurotransmission, as SSR504734 potentiated NMDA-mediated excitatory postsynaptic currents (EPSCs) in rat hippocampal slices (minimal efficacious concentration (MEC): 0.5 μM) and intrastriatal glycine-induced rotations in mice (MED: 1 mg/kg i.p.). It normalized activity in rat models of hippocampal and PFC hypofunctioning (through activation of presynaptic CB1 receptors): it reversed the decrease in electrically evoked [3H]acetylcholine release in hippocampal slices (MEC: 10 nM) and the reduction of PFC neurons firing (MED: 0.3 mg/kg i.v.). SSR504734 prevented ketamine-induced metabolic activation in mice limbic areas and reversed MK-801-induced hyperactivity and increase in EEG spectral energy in mice and rats, respectively (MED: 10–30 mg/kg i.p.). In schizophrenia models, it normalized a spontaneous prepulse inhibition deficit in DBA/2 mice (MED: 15 mg/kg i.p.), and reversed hypersensitivity to locomotor effects of d-amphetamine and selective attention deficits (MED: 1–3 mg/kg i.p.) in adult rats treated neonatally with phencyclidine. Finally, it increased extracellular dopamine in rat PFC (MED: 10 mg/kg i.p.). The compound showed additional activity in depression/anxiety models, such as the chronic mild stress in mice (10 mg/kg i.p.), ultrasonic distress calls in rat pups separated from their mother (MED: 1 mg/kg s.c.), and the increased latency of paradoxical sleep in rats (MED: 30 mg/kg i.p.). In conclusion, SSR504734 is a potent and selective GlyT1 inhibitor, exhibiting activity in schizophrenia, anxiety and depression models. By targeting one of the primary causes of schizophrenia (hypoglutamatergy), it is expected to be efficacious not only against positive but also negative symptoms, cognitive deficits, and comorbid depression/anxiety states.
Similar content being viewed by others

INTRODUCTION
The original hyperdopaminergic hypothesis of schizophrenia (based on the seminal work by Carlsson and Lindqvist (1963) and Seeman et al (1976)) has been more recently complemented by the hypoglutamatergic hypothesis. This latter has been presented by several authors (Javitt and Zukin, 1991; Olney and Farber, 1995) as being more appropriate to explain some aspects of schizophrenia (such as negative symptoms and cognitive deficits) that could not simply be accounted for by the hyperdopaminergic hypothesis. The most robust argument in favor of the hypoglutamatergic hypothesis is the observation that phencyclidine-like compounds (PLC) are able to induce psychoses in human volunteers (Allen and Young, 1978) and to precipitate psychotic episodes in stabilized schizophrenic patients (Lahti et al, 1995; Tamminga et al, 1995). PLC are known to bind to a site localized inside the ionophore of the N-methyl-D-aspartate (NMDA) receptorial complex (Javitt and Zukin, 1991), and as such reduce the NMDA receptor-mediated glutamatergic transmission.
Another, although more indirect, argument for this hypoglutamatergic hypothesis is based on the observation that compounds that putatively potentiate the glutamatergic neurotransmission seem to provide additional benefit when associated with antipsychotics. For example, recent studies, some of them double blind and placebo-controlled, have shown that d-cycloserine, glycine, and d-serine improve some aspects of negative symptomatology and cognitive dysfunction, do not interfere with the beneficial effects of antipsychotics on the positive symptomatology and are also well tolerated (Goff et al, 1995, 1999; Tsai et al, 1998; Heresco-Levy et al, 1998, 1999, 2002; Javitt et al, 2001; Evins et al, 2002). Beneficial effects on negative symptomatology and cognitive dysfunction are of interest, as current antipsychotics are marginally effective against these two aspects of the pathology. Furthermore, cognitive impairment, with its cohort of deficits affecting various dimensions of memory, is nowadays considered to be a core feature of schizophrenia, possibly triggering the positive symptoms (Lewis, 2004). Due to the pivotal role of the glutamatergic system in memory and cognitive processes (Riedel et al, 2003), a compound that would potentiate this system might tackle the pathology at its very roots.
D-Cycloserine, glycine, and d-serine are compounds that act as agonists at the glycine B site that is part of the NMDA receptor. Glycine has been shown to act as a requisite co-agonist of glutamate for activation of this receptor (Johnson and Ascher, 1987). Thus, the strength of the glutamatergic neurotransmission is tightly controlled by the synaptic concentration of glycine in the vicinity of NMDA receptors, so that the more glycine, the stronger the glutamatergic neurotransmission. It has traditionally been assumed that because of the high concentration of synaptic glycine (3–10 μM; see the microdialysis section in Results; Hashimoto and Oka, 1997), the glycine B site is probably saturated. However, recent studies have cast doubt on this assumption and it now appears that it is probably not the case (Bergeron et al, 1998; Chen et al, 2003). Levels of synaptic glycine are tightly controlled by two types of specific transporters (glycine transporters, GlyT1 and GlyT2). GlyT1, classically considered to be localized on glial cells—but just recently described on neurons as well (Cubelos et al, 2005)—is closely associated with the NMDA receptor, whereas GlyT2 is colocalized with inhibitory (ie strychnine-sensitive) glycine receptors (see for review Gomeza et al, 2003). By increasing synaptic concentration of glycine in the vicinity of NMDA receptors, blockers of GlyT1 are expected to potentiate glutamatergic transmission, and as such represent promising targets for pharmacological intervention against schizophrenia (Vandenberg and Aubrey, 2001; Millan, 2002; Slassi and Egle, 2004). Several GlyT1 blockers (ALX5407; also known as NFPS, ORG 24461, ORG 24598 and CP 802,079) have been reported to possess the preclinical profile of putative antipsychotics (to cite a few studies, Bergeron et al, 1998; Atkinson et al, 2001; Brown et al, 2001; Harsing et al, 2003, Kinney et al, 2003, Le Pen et al, 2003, Martina et al, 2004), but data for each compound are generally scant and/or scattered across several papers, and none of these compounds seems to have been developed clinically.
We have recently synthesized the piperidinebenzamide derivative SSR504734 (2-chloro-N-[(S)-phenyl[(2S)-piperidin-2-yl] methyl]-3-trifluoromethyl benzamide, monohydrochloride) (Figure 1) as part of a chemical effort aimed at developing a selective and reversible GlyT1 inhibitor (Dargazanli et al, 2003). We presently report on its in vitro binding profile, and on biochemical, electrophysiological, and behavioral data demonstrating that (1) this compound elevates central glycine levels, (2) increased glycine levels have a positive functional impact on central glutamatergic transmission, (3) augmented glutamatergic tone prevents various effects produced by a hypoglutamatergic state, and (4) SSR504734 shows activity in several models predictive of antipsychotic as well as antidepressant and anxiolytic activities.

Chemical structure of (S,S)SSR504734.
MATERIALS AND METHODS
Animals
Experimental subjects were supplied by Iffa-Credo (Les Oncins, France) or Charles-River (St Aubin-les-Elbeuf, France), unless specified otherwise. Animals were kept in temperature- and humidity-controlled rooms (22°C, 50%) with lights on from 0700 to 1900 (except when indicated otherwise), with water and food available ad libitum. All experiments were performed in accordance with the ‘Guide and Care and Use of Laboratory Animals’ (National Institutes of Health) and were approved by the Animal Ethics Committee of Sanofi-Synthelabo Recherche.
Drugs
SSR504734, the (S,S) enantiomer, SSR506204, the (R,R) enantiomer of SSR504734, ALX5407 (N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy) propyl]sarcosine; NFPS), and phencyclidine were synthesized by the CNS Medicinal Chemistry Department of Sanofi-Synthelabo Recherche. Ketamine, WIN55212-2, sarcosine, glycine, and MK-801 were obtained from Sigma Aldrich (Saint Quentin Fallavier, France) or Bio-Rad Laboratories (Life Science Group, Marnes-la-Coquette, France); d-amphetamine was obtained from Boyer (Paris, France).
Drugs were diluted in saline or distilled water alone or with a few drops of Tween 80, unless specified otherwise. Doses refer to the weight of the free base, except when indicated otherwise; all drug solutions were prepared fresh daily and injected i.p. (10 or 20 ml/kg in mice, 2 or 5 ml/kg in rats), s.c. (0.1 ml in rat pups), or i.v. (1 ml/kg in rats). For p.o. administrations in mice (60 min pretest), SSR504734 was suspended in distilled water with a few drops of Tween 80, and the volume of administration was 10 ml/kg.
Note: ALX5407 (considered by some authors as producing an essentially irreversible blockade of GlyT1; Atkinson et al, 2001; Aubrey and Vandenberg, 2001) was used as a comparator GlyT1 inhibitor solely in tests susceptible to give an indication of the degree of reversibility of the blockade of glycine uptake.
Effects of SSR504734 and Its (R,R) Enantiomer SSR506204 on In Vitro [14C]glycine Uptake
Human neuroblastoma (SK-N-MC) and rat astrocytoma (C6) cell lines, obtained from the American Type Culture Collection (Rockville, MD, USA), were maintained at 37°C, in humidified air with 5% CO2, in monolayer culture in growth medium (MEM for SK-N-MC cells and HAM-F12 for C6 cells) containing 10% fetal calf serum. Cells, collected by trypsination, were subcultured twice a week. The presence of GlyT1 in C6 cells has been reported by Gomeza et al (1995). In SK-N-MC cells, the presence of GlyT1 was assessed as follows: total RNA of cells was isolated using Trizol reagent, and then reverse-transcribed and submitted to PCR amplification (Invitrogen, Carlsbad, USA; initial denaturing step at 93°C for 1 min, and 35 cycles consisting of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min) in the presence of two primers specific for one of the three known GlyT1 isoforms (Kim et al, 1994). Sense primers were 5′-TGC CAA AGG GAT GCT GAA TG-3′ for isoform a and 5′-GCG GCT CAT GGA CCT GTG-3′ for both isoforms b and c. The antisense primer sequence, common to the three isoforms, was 5′-CGC AGA TGA GCA TGA TG-3′. For confirmation, RT-PCR products were analyzed by Southern blot using a 32P-labeled internal oligonucleotide probe specific for each human GlyT1 isoform. Blots were scanned with a Storm Phosphoimager (BD Biosciences, Franklin Lakes, USA).
At 48 h before [14C]glycine uptake experiments, cells were plated at a density of 20 000–30 000 per well in 96-well culture dishes previously coated with fibronectin for SK-N-MC cells or poly-D-lysine for C6 cells. Assays were performed at 37°C in 200 μl of HEPES buffer (HB) containing (in mM) NaCl (147), KCl (5), MgCl2 (2), CaCl2 (2), HEPES (10), D-glucose (10), and L-alanine (5) with pH 7.4. Growth medium was removed and after washing with HB, cells were incubated for 10 min with test compounds. Uptake was started by adding 10 μM [14C]glycine (112.4 mCi/mmol; NEN Life Science Products, Paris, France). Nonspecific uptake was determined with 10 mM unlabeled glycine. After 10 min, cells were washed twice with HB, and scintillation fluid (Optiphase supermix, Wallac, Perkin Elmer, Life Sciences, Courtaboeuf, France) was added to the wells. Radioactivity was measured by liquid scintillation in a Wallac MicroBeta counter.
Results are expressed as the drug concentration required to inhibit 50% (IC50) of specific [14C]glycine uptake, and were obtained by nonlinear regression analysis.
Receptor Selectivity of SSR504734
Interaction of SSR504734 with about 120 different receptors, ion channels, enzymes, or transporters was evaluated by use of standard protocols (Cerep, Celle l'Evescault, France) or by internal studies.
Effects of SSR504734 and SSR506204 on Ex Vivo [14C]glycine Uptake
Male OF1 mice (20–25 g) were killed 30 or 60 min after i.p. or p.o. administration, respectively. Cortical tissues were rapidly dissected and homogenized on ice using a polytron apparatus (1 g tissue for 10 volumes of cold HB). The assay performed on fresh homogenates was started by adding 10 μM [14C]glycine to 20 μl of tissue in HB (final protein concentration: 0.8–0.9 mg/ml). Nonspecific uptake was determined with 10 mM unlabeled glycine. The uptake was performed for 10 min at 25°C, and stopped by aspiration onto MultiScreen glass fiber filter plate using a MultiScreen vacuum manifold. The filter was washed twice with ice-cold HB, dried, and soaked with scintillation fluid (Meltilex, Wallac, Perkin Elmer, Life Sciences, Courtaboeuf, France). Radioactivity was measured by liquid scintillation in a Wallac MicroBeta counter.
A time-course study was performed at 5 min, 15 min, 1 h, 4 h, 7 h, 16 h, and 24 h after oral treatment with 30 mg/kg of SSR504734, using the protocol described above.
Results are expressed as the percentage of [14C]glycine uptake vs the control (vehicle-treated) group and as the dose of the compound that inhibits 50% of uptake (ID50) calculated by nonlinear regression analysis. For the time-course experiment, data were analyzed with a one-way ANOVA, followed by post hoc Dunnett's tests.
Effect of SSR504734 on Extracellular Levels of Glycine Measured in the Prefrontal Cortex of Freely Moving Rats
Male Sprague–Dawley rats (280–330 g) were housed two per cage. At 2 days before the dialysis measurements, they were anesthetized with chloral hydrate (400 mg/kg i.p., 10 ml/kg of body weight) and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). Anesthesia was maintained throughout surgery as necessary with supplementary doses of chloral hydrate. Body temperature was monitored by a rectal probe and adjusted (37±1°C) by a homeothermic blanket. The skull and the dura were opened to allow the implantation of a guide cannula in the medial prefrontal cortex (PFC). The coordinates were 2.5 mm anterior to bregma, 0.6 mm lateral to the midline, and 1.3 mm below the dural surface (Paxinos and Watson, 1998). A dental cement cap held the cannula in place, and three screws anchored the cap to the skull. The rats were individually housed postsurgery and allowed 2 days of recovery before the start of the experiment. On the day of the experiment, animals were placed in a microdialysis bowl, the obturator of the cannula was removed, and a 3 mm microdialysis probe (CMA12, Carnegie Medicine AB, Stockholm, Sweden) was inserted into the guide cannula. The probe was perfused at a constant flow rate of 1 μl/min using a microinjection pump (CMA100; Carnegie Medicine AB) with a gassed Ringer's solution containing (in mM) NaCl (145), KCl (2.7), CaCl2 (1.2), MgCl2 (1), Na2HPO4 (2.3), and NaH2PO4 (0.45); pH 7.4. Microdialysis sampling started 120 min after probe placement into the PFC. The outlet of the probe was connected to an online derivatization system as described previously (Bert et al, 1996), allowing direct analysis of dialysate samples collected every 15 min.
Glycine levels were measured in 15 μl dialysate samples using capillary electrophoresis (CE) with laser-induced fluorescence detection. Before analysis, the samples were derivatized using naphtalene-2,3-dicarboxaldehyde and sodium cyanide, as described previously (Bert et al, 1996). CE experiments were performed on a P/ACE MDQ capillary electrophoresis system (Beckman Coulter, Villepinte, France) coupled to an external Zetalif fluorescence detector (Picometrics SA, Toulouse, France). The excitation was performed by an Omnichrome (Melles Griot Laser Products) helium–cadmium laser at a wavelength of 442 nm with a 30 mW excitation power. The emission intensity was measured at a wavelength of 490 nm. Separations were carried out with a fused-silica capillary (Polymicro Technology, Phoenix, AZ, USA) of 50 μm i.d. and 375 μm o.d. having a total length of 55 cm and an effective length of 38.9 cm with an applied voltage of 25 kV (ie 65 μA current). Borate buffer (75 mM) containing β-cyclodextrin (1 mM), pH 10.5, was used for CE running.
At the end of the experiments, an injection of sky blue solution was performed through the probe and animals were killed with an overdose of pentobarbital. The brain was removed, frozen, and 50-μm-thick sections were cut with a cryostat to verify correct placement of the microdialysis probe.
Glycine levels in fractional samples were converted to a percentage of the mean value of the 90 min baseline measurements before treatment. Time-course effects of SSR504734 and SSR506204 on glycine levels were analyzed by two-way ANOVAs, with treatment as a between-subjects factor and time of sampling as a within-subjects factor, followed by Dunnett's post hoc tests. Dose effects of SSR504734 were evaluated by comparing the area under the curve during the first 180 min after i.p. injection of the drug or vehicle. Statistical analysis was carried out by a one-way ANOVA followed by Dunnett's post hoc tests.
Effect of SSR504734 on Evoked NMDA-Mediated Excitatory Postsynaptic Currents in Rat Hippocampal Slices
Sprague–Dawley rats (17–22 days old) were killed and their brains removed and sectioned (coronal 0.3-mm-thick slices) with a Campden 752M microslicer in an artificial cerebrospinal fluid (aCSF) solution containing (in mM) NaCl (126), KCl (3), MgCl2 (1), KH2PO4 (1), CaCl2 (1), NaHCO3 (25), and glucose (11), pH 7.35, at approximately 0°C and aerated with 95% O2 and 5% CO2. After at least 1 h in aCSF at room temperature, slices were transferred to the recording chamber and superfused continuously with aCSF at 30–32°C. Patch-clamp whole-cell recordings of layer CA1 pyramidal cells were obtained with borosilicate glass electrodes (resistance: 4–7 MΩ), filled with a 290 mosmol solution containing (in mM) KMeSo4 (130), EGTA (10), ATP (2), GTP (0.5), and QX314 (5); pH 7.2.
Electrical recordings were made from CA1 pyramidal cell somata under visual control with a × 10 water-immersion lens on an upright microscope (BX50WI; Olympus Optical, Tokyo, Japan). A concentric bipolar stimulation electrode (platinum/iridium, 12.5 μm i.d., Frederick Haer and Co., ME, USA) connected to a constant current isolated stimulator (DS3, Digitimer Ltd, UK) was placed near the pyramidal layer of CA1 to generate evoked NMDA-mediated excitatory postsynaptic currents (eEPSCs). The bath solution was supplemented with 20 μM bicuculline methiodide, 1 μM CGP-52432, 1 μM strychnine, and 1 μM NBQX (antagonists for GABAA, GABAB, inhibitory strychnine-sensitive glycine, and AMPA/kainate receptors, respectively) in order to isolate the NMDA receptor-mediated component of eEPSCs. The concentration of external magnesium was maintained at 1 mM to minimize polysynaptic transmission due to high activity of NMDA transmission.
In all experiments, 10 μM of glycine was added to the aCSF medium, experimental conditions that are similar to those used by Chen et al (2003). This extracellular ‘basal’ glycine concentration did not affect the amplitude of eEPSCs but was added to avoid glycine depletion of the permanently superfused slices.
Parameters of stimulation (square pulses, duration 10–100 μs and amplitude 2–10 mA) were adjusted to obtain half-maximal amplitude of monophasic eEPSCs and elicited at a fixed frequency of 0.2 Hz.
Data are expressed as the percentage of the eEPSCs amplitude obtained under control conditions. Statistical significance of drug-induced effects was assessed using paired Student's t-test. For the concentration-dependent curve, a least square fitting method was used with the single-site model equation of nonlinear regression (Levenberg–Marquardt). Parameters providing the best fit are given with a 95% confidence interval.
Effects of SSR504734 and SSR506204 on Contralateral Rotations Induced by Intrastriatal Microinjection of Glycine in Mice
At 1 week before experiments, female CD1 mice (25–27 g) were housed in groups of 10 per cage. Details of the microinjection procedure have been published elsewhere (Poncelet et al, 1993).
SSR504734, SSR506204, or vehicle was injected i.p. 30 min before intrastriatal microinjection of glycine (0.001 ng, a subliminal dose). In another experiment, MK-801 (0.03 mg/kg i.p.) or vehicle was administered 15 min before SSR504734 (1 mg/kg i.p.) or vehicle, followed 30 min later by a microinjection of 0.001 ng glycine.
The number of complete contralateral (ie away from the injection side) rotations was visually recorded and cumulated over three periods of 2 min (2–4, 5–7, and 8–10 min after microinjection of glycine). Statistical analysis was performed using one-way ANOVAs, followed by Dunnett's post hoc tests.
Note: Presynaptic CB1 receptors are known to negatively regulate excitatory synaptic transmission, presumably through reduced release of neurotransmitter such as glutamate, notably in the hippocampus and PFC (Gifford and Ashby, 1996; Auclair et al, 2000; Schlicker and Kathmann, 2001). This alternative approach for producing a hypoglutamatergic tone was exploited in the following two experiments.
Effects of SSR504734 and SSR506204 on the Decrease of [3H]acetylcholine Release Induced by WIN55212-2 in Rat Hippocampal Slices
Male OFA rats (250–350 g) were decapitated, their brains quickly removed, and the hippocampus dissected on an ice-cold aluminum block. Following dissection, 350 μm tissue slices were cut with a vibratome, and the slices transferred to 20 ml of Krebs buffer (pH 7.4) saturated with 95% O2 and 5% CO2 and containing 20 μM [3H]choline (86 Ci/mmol). Following incubation with the radiolabel for 30 min at 37°C, slices were transferred to 18 superfusion chambers (three slices per chamber). Slices were superfused at 37°C, at a rate of 0.5 ml/min, with oxygenated Krebs buffer containing 10 μM hemicholinium to prevent the reuptake of [3H]choline. Sample collection was started after a 45 min wash period. Two 3 min fractions were subsequently collected in order to measure basal release. The cannabinoid-1 (CB1) receptor agonist WIN55212-2 (10−6 M) was introduced during the fifth collection period and was present until the end of the experiment. SSR504734 was added 9 min prior to WIN55212-2. Release of [3H]acetylcholine ([3H]ACh) was electrically evoked (rectangular pulses, 2 Hz, 2 ms, 1 mA) for 3 min, after which the original Krebs buffer was introduced to re-establish basal release. Results are expressed as percent of inhibition of controls. Data were analyzed by means of one-way ANOVAs followed by Dunnett's post hoc tests. An IC50 for the effects of SSR504734 was determined with a four-parameter logistic model using a weighed linear curve fitting program.
Effects of SSR504734 and SSR506204 on the Decrease of Firing Rate of Prefrontal Cortex Neurons Induced by WIN55212-2
Male Sprague–Dawley rats (280–400 g) were anesthetized with chloral hydrate (400 mg/kg i.p.) and placed in a stereotaxic apparatus (David-Kopf Instruments, Tujunga, CA, USA). Adequate level of anesthesia was maintained throughout the experiment with supplementary doses of choral hydrate. The animal's temperature was maintained at 37±0.1°C by an electronic heating pad device (Harvard Apparatus, Edenbridge, UK). After appropriate surgery, small burr holes were drilled into the skull bone above the areas to be explored. Extracellular recordings were made with 1-μm-tipped glass micropipettes filled either with 1 M NaCl, or 0.5 M sodium acetate containing 2% Pontamine Sky Blue (impedance 4–7 MΩ at 100 Hz) aimed at medial PFC cells: 2.9–3.2 and 0.6–0.8 mm anterior and lateral to bregma, respectively, and 2.2–3.6 mm below the cortical surface (Paxinos and Watson, 1998). A hydraulic microdrive (MMO-203, Narishige, Tokyo, Japan) allowed fine 3-D electrode movement. Action potentials (spikes) were amplified, filtered (400 Hz–5 kHz), and monitored on a digital oscilloscope and an audio amplifier. The potentials were fed into a window discriminator and analyzed online using a ‘μ1401 Intelligent Laboratory Interface’ connected to a PC running the CED ‘Spike2’ software (Cambridge Electronic Design, Cambridge, UK).
Firing rate histograms were run in a continuous mode (10 s bin size). The firing rate was allowed to stabilize for 10–15 min before the first i.v. injection of SSR504734, SSR506204, or vehicle (50 μl of 1 N HCl+distilled water for 1 ml/kg v/w). WIN55212-2 was administered (0.1 mg/kg i.v.) 10 min after the test drug (or vehicle). The firing rate was then monitored for at least an additional 30 min. A single cell was recorded per rat. All i.v. injections were performed over a duration of 1 min.
Mean firing rates (in spikes/s) were calculated online. Comparison was made between the 5 min periods following and preceding injection of WIN55212-2 (ie those following injection of the test drug or vehicle). The difference between values obtained before and after administration of WIN55212-2 was assessed by means of one-way ANOVAs followed by Duncan's post hoc tests.
Effect of SSR504734 on Extracellular Levels of Dopamine in the Prefrontal Cortex of Freely Moving Rats
Note: The capacity of some atypical antipsychotics to augment dopaminergic neurotransmission in the PFC has been suggested by some authors (Kapur and Remington, 1996) to form the basis for their beneficial impact on negative symptoms and cognitive deficits in schizophrenic patients. For this reason, we also measured the ability of SSR504734 to augment extracellular DA tone in this structure.
Adult male Sprague–Dawley rats (280–300 g) were anesthetized with chloral hydrate (400 mg/kg i.p.) and placed in a stereotaxic frame (David Kop Instruments, Tujunga, CA, USA) and a guide cannula was stereotaxically implanted above the medial PFC (3.5 mm anterior to bregma, 0.5 mm lateral to bregma; Paxinos and Watson 1998). At 24 h after surgery, a microdialysis probe (Carnegie Medicine, Stockholm, Sweden), 250 μm in diameter with an exposed membrane length of 4 mm, was positioned within the guide cannula (vertical coordinates: 5 mm under the cortical surface) and perfused (CMA/100 pump, Carnegie Medicine) at a flow rate of 2 μl/min with aCSF containing (in mM) NaCl (147), KCl (4), CaCl2 (1.2), and MgCl2 (1). The animals were left for at least 3 h to allow the system to equilibrate, and then dialysate samples were collected every 20 min and analyzed using HPLC with electrochemical detection (for details of the methodology, see Curet et al, 1996). The average concentration of four stable fractions immediately preceding i.p. administration of the compound or vehicle was defined as the 100% control value and dopamine (DA) levels in serial perfusates were converted to a percentage of this mean value. Statistical analysis was carried out by a two-way ANOVA (with time as the within factor and treatment as the between factor), followed by one-way ANOVAs at each time, with Dunnett's post hoc tests.
Effect of SSR504734 on Ketamine-Induced Increase of Brain Metabolic Activity in Mice
Male OF1 mice (28–37 g) were housed 10 per cage, 1 week before the experiment. On the day of the experiment, they were housed in individual cages at least 1 h before treatment. Vehicle or SSR504734 was injected i.p. 30 min before administration of ketamine (30 mg/kg i.p.) or saline (ketamine was chosen instead of PCP or MK-801 since it was the compound used in the princeps study by Duncan et al, 1998a). The solution of 2-deoxy-D-[1-14C]glucose (2-DG; Amersham Biosciences, Buckinghamshire, England) was slowly injected (13 μCi in 0.3 ml of saline, over 20 s) into the tail vein 2 min after injection of ketamine or saline. Mice were killed by decapitation 5 min after the i.v. injection of 2-DG. Brains were removed and frozen by immersion in cooled isopentane (−45°C), and then stored at about −80°C until sectioned.
Cryostat coronal sections (20 μm) were thaw-mounted onto microscope coverslips, apposed to Kodak Biomax MR film (Amersham, Fontenay-sous-Bois, France) along with a set of 14C standards calibrated for equivalent 14C concentrations in 20-μm-thick brain sections (Amersham, France), and stored in X-ray cassettes for an exposure period of 2 days. Films were developed manually (5 min in developer LX24 and 5 min in X-ray fixer AL4; Kodak, France).
Optical densities in autoradiograms were quantified by digital image analysis, with a video-based system (Densirag, Version D2.99, Biocom, Les Ulis, France). Regional brain identification was made according to the atlas of Franklin and Paxinos (1997) for mice. Amounts of radioactivity were derived by relating the optical density in brain regions to those of 14C standards. For each brain region (prefrontal and cingulate cortices, nucleus accumbens, anteroventral thalamic nucleus, dorsal hippocampus, and fimbria), 3–6 sections per animal were analyzed and 14C tissue concentration values were averaged. Relative 2-DG uptake data are expressed as the ratio of tissue 14C concentration in gray matter regions relative to that in the fimbria (taken as reference), and the resulting values multiplied by 100. For each brain region, relative 2-DG uptake values were analyzed with a one-way ANOVA, followed by Newman–Keuls post hoc tests.
Effects of SSR504734 and SSR506204 on Locomotor Hyperactivity Induced by MK-801 in Mice
Male Swiss mice (18–21 g) were individually isolated in boxes and pretreated i.p. with SSR504734, SSR506204, or vehicle, immediately followed by vehicle or MK-801 (0.2 mg/kg i.p.). At 30 min after the second injection, they were placed in actimeters (20 cm diameter, 9.5 cm height; Apelex, France) equipped with two perpendicular light beams 1.5 cm above the floor. Locomotor activity (number of interrupted light beams) was recorded for a period of 30 min after placing the mouse into the actimeter. Data were analyzed with one-way ANOVAs, followed by Dunnett's post hoc tests.
Effect of SSR504734 on the Increase of the Alpha1 Band Spectral Energy Induced by MK-801 in Rats
Male Sprague–Dawley rats (220–240 g) were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and mounted in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). Cortical electrodes (small stainless-steel screw 0.9 mm in diameter) were screwed into the bone at the level of the sensorimotor cortex (1.5 mm lateral to the median suture and 1.5 mm behind the fronto-parietal suture), the visual cortex (1.5 mm lateral to the median suture and 1.5 mm in front of the parieto-occipital suture), and the cerebellum (reference electrode). Cortical electrodes were soldered to a miniature connector fixed with dental cement to the cranium.
After 3 weeks of postoperative recovery, animals were placed in Plexiglas cylinders (60 cm diameter) with free access to food and water. For studying the effect of SSR504734 on the increase of the alpha1 spectral energy induced by MK-801, after 15 min of EEG recording (first period: control), vehicle or SSR504734 was administered i.p., followed 15 min later by MK-801 (0.2 mg/kg i.p.). EEG signals were amplified and filtered (1–100 Hz). Sequential spectral analysis was performed automatically by means of a computerized system (‘Coherence 32’ software, Deltamed, Paris, France). Signals were sampled (128 Hz) for the Fourier transformation, which allowed calculation of the power variable (μV2). Absolute power spectra of EEG signals were computed every 30 s from 0.5 to 32 Hz in steps of 0.5 Hz and were calculated for each of the following frequency bands: delta band (0.5–3.5 Hz), theta band (theta1: 4–6 Hz; theta2: 6.5–8 Hz), alpha band (alpha1: 8.5–11 Hz; alpha2: 11.5–13.5 Hz), and beta band (14–32 Hz). MK-801 was tested at 0.2 mg/kg i.p., because it induced a large increase of the absolute power within the alpha1 band at this dose.
The EEG spectral power of the alpha1 band was averaged for 15 min before administration of MK-801, and every 10 min for 1 h after injection of MK-801. Drug-induced changes were evaluated by calculating the ratio of the absolute power after i.p. injection of SSR504734 or vehicle on the absolute power during the control period (taken as 100%) and expressed as percent of the power during the control period.
The results are expressed as percentages of the control values. Statistical analysis was carried out using a two-way ANOVA (with treatment as the between-subjects factor and periods of recording as the within-subjects factor), followed by one-way ANOVAs (and post hoc Dunnett's tests) with treatment as the between factor, for each of the six 10 min recording periods.
Effect of SSR504734 on the Impairment of Novelty Discrimination in Adult Rats Treated with Phencyclidine at the Neonatal Stage
Female Wistar Han rats were obtained with 10 male pups on postnatal day 3 (PN 3). Pups were treated on PN 7, 9, and 11 with 10 mg/kg of PCP (s.c. administration, 1 ml/100 g body weight) or vehicle. Pups from the same litter received an identical treatment. The mother and pups were housed together until weaning at PN 21, at which stage pups where housed five per cage until 2 weeks before the beginning of behavioral experiments, when they were housed individually. Behavioral experiments were performed once they reached the adult stage (between PN 60 and PN 108).
Juvenile male Wistar rats (3 weeks old, 45–50 g on arrival) were housed five per cage. They were left alone for 1 week, before the beginning of experiments (presentation to the adult rats treated at the neonatal stage; see above) that lasted for a week. Each juvenile was used only once a day, and was chosen at random as first or second for presentation to the adult. All animals (mothers, pups, adults, and juveniles) were kept on a reversed light–dark cycle (light on from 1900 to 0700).
Experiments were performed during the dark phase, under infrared illumination (15 lux). Juvenile rats were isolated 30 min before being placed into the home cage of an adult rat. The cage was placed underneath a video camera and the mesh top was removed and replaced by a Plexiglas cover. A first (familiar) juvenile was placed inside the home cage containing one adult rat for a period of 30 min. A second (novel) juvenile was introduced at the end of this period. Durations of investigation behavior (nosing, sniffing, grooming, close chase of the juvenile rat) between the adult rat and each of the two juveniles were recorded manually for a period of 5 min following the introduction of the novel juvenile, by an observer located in an adjacent room fitted with a video monitor. SSR504734 or vehicle was administered i.p. to the adult rat 30 min before exposure to the first juvenile. Each adult rat was subjected to four treatments: one vehicle and three doses of SSR504734. Treatments were administered with 1 or 2 days between each treatment.
Data are expressed as the mean of a novelty discrimination index (NDI), which was calculated as the ratio of the time spent investigating the novel juvenile divided by the time spent investigating the familiar juvenile. NDIs were first log-transformed because of the limited number of subjects and the lack of homogeneity of variances between groups. Statistical analysis was carried out using a two-way ANOVA for repeated measures, with treatment at the neonatal stage as the between-subjects factor and acute treatment at the adult stage as the within-subjects factor, followed by appropriate post hoc tests.
Effect of SSR504734 on the Hypersensitivity to an Acute Challenge with d-Amphetamine in Adult Rats Treated with Phencyclidine at the Neonatal Stage
Adult rats used in this experiment were those used in the novelty discrimination experiment. Animals were first placed into the experiment room 1 h before the experiment was started. They were then placed in activity chambers (Plexiglas boxes 40 × 40 × 30 cm; Digiscan, AccuScan Instruments Inc., Columbus, USA), fitted with infrared beams (2.5 cm apart, 2.5 cm above the floor). Their basal locomotor activity was first evaluated during 30 min (habituation period). At the end of this period, they were injected i.p. with SSR504734 or vehicle, and replaced into the actimeters. After 30 min, they were injected i.p. with d-amphetamine (2 mg/kg) or vehicle, and their locomotor activity was measured during 90 min. Each rat was subjected to five treatments: two vehicle (alone and with d-amphetamine) and three doses of SSR504734 in combination with d-amphetamine. Treatments were administered with a minimum of 3 days between each treatment.
Data are expressed as the mean number of infrared beam interruptions, and were analyzed with a two-way ANOVA for repeated measures, with treatment at the neonatal stage as the between-subjects factor and acute treatment at the adult stage as the within-subjects factor, followed by appropriate post hoc tests.
Effect of SSR504734 on a Spontaneous Deficit of Prepulse Inhibition of the Startle Reflex in DBA/2 Mice
Male DBA/2J mice (22–26 g) were housed in groups of four per cage. They were subjected to a selection session (see below) 13 days after their arrival. Mice were tested in six startle boxes (SR-LAB, San Diego Instruments, San Diego, CA). The startle reflex was detected and transduced by a piezoelectric accelerometer positioned underneath the startle platform, and digitized and stored by a microcomputer running the SR-LAB software (that also controlled all other events). Mice were first placed inside a Plexiglas restraint cylinder (3.7 × 12.8 cm long) fixed on top of the startle platform. The selection session was then started: after 5 min of habituation, 20 stimuli (separated by a variable interval: 5–25 s, by steps of 5 s) were delivered—17 startling pulses stimuli (P; 40 ms duration, 120 dB intensity) intertwined with three prepulses at 30 dB followed by a pulse stimulus (p30/P). For these last three stimuli, prepulses were of 20 ms duration and 88 dB (that is 30 dB above background noise of 58 dB), and pulses were as above, with 100 ms between the end of the prepulse and the beginning of the pulse. Audio stimuli were calibrated (±2 dB, A scale) with a sound level meter (model CDA 830, Chauvin Arnoux, France). Startle platforms were calibrated (SR-LAB calibrator standardization unit), with less than 3% variation between platforms. The startle reflex was recorded during the 100 ms following onset of the startling stimulus (sampling interval: 1 ms). Startle amplitude was defined as the peak amplitude of the downward force exerted by the startled mouse on the platform. Throughout the experiment, mice were in the dark. This selection session was used to distribute mice across groups with similar levels of basal startle for subsequent testing.
On the second day (test session), mice were injected i.p. with vehicle or SSR504734, 30 min before being placed into the restraint cylinders. The test session started with a 5 min habituation period, followed by delivery of four pulses alone (P). These were followed by two identical blocks of stimuli: each block comprised a sequence of 26 stimuli, divided into five conditions: 10 startle pulses without prepulses (P), four startle pulses preceded by 6 dB prepulses (p6/P), four startle pulses preceded by 18 dB prepulses (p18/P), four startle pulses preceded by 30 dB prepulses (p30/P), and four stimuli with no prepulses and no pulses (nop/noP). The five conditions were delivered in a pseudo-random order. Otherwise, all other parameters were similar to those in effect for the selection session described above.
Prepulse inhibition (PPI) for each of the three prepulse intensities was calculated for each mouse from averaged peak startle amplitudes obtained during presentation of the 26 stimuli presented during block 1 (data from block 2 were not analyzed). Data from the four P's delivered following the acclimation period were not used. PPIs (expressed as percentage) were calculated as follows:

PPIs were analyzed by means of a two-way ANOVA for repeated measures (between subjects factor: drug dose; within subjects factor: intensity of prepulse stimuli), followed by a Winer analysis at each level of PPI. In addition, effects of drugs on basal startle (P stimuli; startling pulses only) amplitude were analyzed using a one-way ANOVA with the dose as the between factor, followed by Dunnett's post hoc tests.
Effects of SSR504734 and SSR506204 on Ultrasonic Distress Vocalizations in Rat Pups
Female Sprague–Dawley rats were obtained with 10 male pups on PN 3–4. The procedure, adapted from the one described by Gardner (1985), was as follows: each pup (PN 7) was first separated from its mother and littermates, injected s.c. (0.1 ml) with SSR504734 or vehicle, and returned to its mother. After 30 min, the pup was placed in a soundproof cage. The Ultravox system (Noldus, Wageningen, The Netherlands) was used to record ultrasonic vocalizations (UV, in the 40 kHz range). First, a modified ultrasound detector (Mini-3 bat model) connected to an electret microphone (positioned next to the pup) was used to transform ultrasonic sound into audible sound. The signal was then filtered (user-defined frequency range and amplitude threshold) and sent to a PC, where the UltraVox software recorded each bout of ultrasonic vocalizations during the 3 min test session.
Data are presented as the mean number of ultrasonic calls lasting more than 10 ms, and were analyzed with a one-way ANOVA, followed by Dunnett's post hoc tests.
Effect of SSR504734 in a Chronic Mild Stress Procedure in Mice
The chronic mild stress (CMS) protocol, originally described by Willner et al (1992) for rats, was adapted from the one described by Kopp et al (1999) for mice. It consisted in the sequential application of a variety of mild stressors, including restraint, forced swimming, water and/or food deprivation, pairing with another stressed animal, each applied for a period ranging from 2 to 24 h, in a schedule lasting 3 weeks, which was repeated as necessary until the end of the experiment.
Administration of SSR504734 (10 mg/kg i.p.) was started 2 weeks after the beginning of the CMS. The dose chosen was one that had been shown to have efficacy in mice in neurochemical and behavioral models. Animals (BALB/c male mice, 21–28 g at the start of the experiment) were injected i.p. once a day until all experiments were completed (33 days).
Parallels between depression in humans and the behavior of chronically stressed animals have been drawn on the difficulty of the patient to accomplish even the smallest tasks (eg washing and dressing in the morning), leading to the inability to maintain minimal personal hygiene, and the decrease in grooming behavior seen in chronically stressed animals. In this latter case, there is a degradation of the physical state of the coat, consisting mainly in dirtying and/or loss of fur. Based on these observations, we measured physical state of the coat once a week over the entire CMS period, using the following scale:
3: clean and well-groomed coat
2: disorganized (poorly groomed) coat on the back
1: dirty coat with loss of patches of fur
Data are presented as the mean physical state scores, and were analyzed with a two-way ANOVA (with treatment as the between-subjects factor and time (week of treatment) as the within-subjects factor), followed by one-way ANOVAs and post hoc Newman–Keuls tests, with treatment as the between factor, for each of the 7 weeks of CMS.
Effects of SSR504734 and SSR506204 on the Sleep/Wakefulness Cycle in Freely Moving Rats
Details of the surgical procedure were as described in ‘Effect of SSR504734 on the Increase of the Alpha1 Band Spectral Energy Induced by MK-801 in Rats’.
Recording sessions took place from 1100 to 1700 during 3 consecutive days: precontrol (Day 1), drug challenge (Day 2), and postcontrol (Day 3). EEG activity in sensorimotor and visual cortices was recorded with the cerebellar electrode as the reference. Three stages were differentiated: wakefulness (W; characterized by low-voltage EEG activity), slow-wave sleep (SWS; characterized by an increase in EEG activity, that is, high-amplitude slow waves with some bursts of sleep spindles), and paradoxical sleep (PS; characterized by hypersynchronization of the theta rhythm in the visual cortex). Analysis of the EEG signal was performed automatically by means of a computerized system discriminating between the various sleep phases using spectral frequency analysis (‘Coherence 32’ software, Deltamed, Paris, France). Vehicle (Days 1 and 3), SSR504734, or SSR506204 (Day 2) was administered i.p. 15 min before recording. The effects of compounds on the time spent in W, in SWS, in PS, the number of episodes of PS, and the latency time to enter PS were analyzed over a 6 h period and are expressed as the percentage of the control values obtained on Day 1. For each treatment, statistical analysis was carried out using one-way ANOVAs for repeated measures on raw values, followed by Dunnett's post hoc tests (Day 1 as control).
RESULTS
Blockade by SSR504734 of In Vitro [14C]glycine Uptake
Southern blot analysis of RT-PCR products revealed that human SK-N-MC cells only expressed the GlyT1 mRNA isoform a. The other GlyT1 mRNA isoforms b and c were not detected in this cell line (data not shown). [14C]glycine uptake in SK-N-MC cells was saturable, sodium- and chloride-dependent (data not shown), and exhibited a Km=21±4 μM and a Vmax=667±186 pmol/min/106 cells (n=4 independent experiments). Specific uptake was typically 90% of total radioactivity. This is consistent with characteristics described previously for these transporters (Mallorga et al, 2003).
SSR504734 inhibited [14C]glycine uptake in human SK-N-MC and rat C6 cells, with IC50 values of 18±6 and 15±2 nM, respectively (Table 1). The (R,R) enantiomer SSR506204 was approximately 10-fold less potent in blocking the uptake of [14C]glycine, both in human and rat GlyT1.
The IC50 for the reference GlyT1 inhibitor sarcosine in human SK-N-MC cells was 60±10 μM, a value that is almost identical to that previously reported for this compound in other human cells (Herdon et al, 2001).
SSR504734 was inactive (inhibition lower than 50% at 1 μM) against ca 120 targets, including glycine, glutamate, DA, 5-HT, adrenaline, noradrenaline, histamine and muscarinic receptors, enzymes such as MAO, and uptake systems such as DA, 5-HT and noradrenaline transporters (assays performed by Cerep, Celle l'Evescault, France, data available upon request). SSR504734 also had no effect (IC50>1 μM) on human GlyT2 and D-serine transporters, and on murine proline, glutamate, and GABA transporters (data available upon request).
Blockade by SSR504734 of Ex Vivo [14C]glycine Uptake in Mouse Cortical Homogenate
Peripheral administration of SSR504734 produced a dose-dependent inhibition of ex vivo-specific [14C]glycine uptake in the mouse cerebral cortex. ID50 values were 5.0 and 4.6 mg/kg for i.p. and p.o. treatments, respectively, with the p.o./i.p. ratio close to unity, indicating good oral bioavailability (Figure 2a). The (R,R) enantiomer SSR506204 was more than 10 times less potent (ID50: 85.3 mg/kg i.p.; curve not shown) than SSR504734 in blocking the uptake of [14C]glycine in this model.

(a) Dose-dependent blockade by SSR504734 of the ex vivo uptake of [14C]glycine in mouse cortical homogenate. (b) SSR504734 (30 mg/kg p.o.) produced a rapid, long-lasting but reversible blockade of ex vivo uptake of [14C]glycine in mouse cortical homogenate. For both panels, each point represents the mean and SEM of percentages of specific [14C]glycine uptake compared to the control (vehicle-treated) group. Injection times were 30 min (i.p.) and 60 min (p.o) before killing. **P<0.01, compared to the control group (Dunnett's post hoc test following a one-way ANOVA). N=4–6 mice per group, four determinations per mice.
A time-course study, performed at 30 mg/kg p.o., showed that SSR504734 induced a rapid (within 30 min) and significant (F(7,33)=34.55, P<0.0001) decrease of specific [14C]glycine uptake, which was maintained at about 80% inhibition from 1 to 7 h after administration (Figure 2b). The inhibition of specific [14C]glycine uptake was reversible, and values recorded at 16 and 24 h returned to control levels. By contrast, blockade of specific [14C]glycine uptake by ALX5407, at 10 mg/kg p.o., was still maximal (ca 80% inhibition) at 24 h after administration, suggesting that this blocking effect was irreversible (data not shown).
Increase by SSR504734 of Extracellular Levels of Glycine in the Prefrontal Cortex of Freely Moving Rats
The average basal extracellular glycine level in the PFC was estimated to be 3.1±0.3 μM (n=33). Administration of SSR504734 (10 mg/kg i.p.) produced a rapid and sustained increase in PFC extracellular levels of glycine (F(1,16)=10.23, P<0.01 and F(12,192)=8.03, P<0.001 for the treatment and treatment × time interaction factor, respectively; Figure 3a). This effect reached a statistical significance between 45 and 180 min after SSR504734 injection (maximal increase observed at 90 min, 99±21% of control). When tested under similar conditions, the (R,R) enantiomer SSR506204 (10 mg/kg i.p.) was without effect on cortical glycine concentrations (data not shown). Areas under the curve during the 180 min after drug injection indicate that the effect of SSR504734 was dose-dependent (F(3,30)=4.62, P<0.01; Figure 3b). Additionally, SSR504734, at the same dose range, did not increase levels of other amino acids such as glutamate, taurine, alanine, and d-serine (data not shown).

Increase by SSR504734 of extracellular levels of glycine in the PFC of freely moving rats. (a) Time course of the effect of SSR504734. Changes in glycine levels are expressed as a percentage of the mean value of the six basal samples collected before the start of the treatment (indicated by a vertical arrow). Each symbol represents the mean+SEM. **P<0.01, compared with the vehicle group at the corresponding time of sampling (Dunnett's post hoc tests following a two-way ANOVA). N=9 rats per group. (b) Dose-effect relationship of SSR504734. Each bar represents the mean+SEM of the area under the curve (AUC) for the 180 min period following injection of SSR504734 or vehicle. (*)P=0.06, **P<0.01, compared with the vehicle group (Dunnett's post hoc tests following a one-way ANOVA). N=8–9 rats per group.
Potentiation by SSR504734 of Evoked NMDA-Mediated Excitatory Postsynaptic Currents in Rat Hippocampal Slices
The experimental conditions that were chosen allowed us to record only the NMDA receptor component of pyramidal neuron eEPSCs (see Materials and methods) after electrical stimulation within the CA1 area. This was confirmed by the strong voltage dependence (due to the magnesium block) of the eEPSCs (average peak current: 78 pA at −20 mV; Figure 4a) and the full block obtained by bath application of 100 μM 5,7-DCK (or 50 μM L-AP5; data not shown), two antagonists acting at the glycine recognition site and glutamate binding site of the NMDA receptor, respectively (Figure 4b, inset).

Potentiation by SSR504734 of eEPSCs in rat hippocampal slices. (a) Normalized NMDA-mediated eEPSCs (maximum peak amplitude) evoked at different holding potentials in the presence of 1 mM [Mg2+]out and 10 μM [glycine]out. The graphical plot of peak eEPSCs displays a region of negative slope conductance, characteristic of NMDA-mediated eEPSCs. N=3 cells per point. Inset: Single traces of eEPSCs from −70 to +40 mV (step: 10 mV). (b) Concentration-dependent ratio increase produced by SSR504734 (squares) or SSR506204 (triangle). Each symbol represents the mean±SEM ratio change of maximal amplitudes of eEPSCs obtained against a background concentration of glycine (10 μM). Experiments were realized at Vh=−20 mV, to maximize NMDAR-mediated currents. *P<0.05, **P<0.01 with respect to control condition (10 μM glycine alone); paired Student's t-tests. Numbers of cells per group are given in parentheses. Inset: Single traces of eEPSCs showing concentration-dependent potentiating effects of SSR504734, and blockade by the glycine/NMDA site antagonist 5,7 DCK.
SSR504734 increased the amplitude of eEPSCs in a concentration-dependent manner (Figure 4b), with an EC50=0.67±0.21 μM and a maximal enhancement of 132±0.27% at 3 μM as compared to the control amplitude. The less active enantiomer SSR506204 (Figure 4b, triangle) had no effect at 3 μM, a concentration at which SSR504734 produced a more than two-fold increase of eEPSCs.
The increase in eEPSCs amplitude observed with SSR504734 was reversible within 8 min upon removal of the compound. Under similar conditions, ALX5407 (0.3 μM) also potentiated eEPSCs (41±8% vs control conditions) but this potentiating effect appeared to be irreversible, since it was still present 20 min after washing.
Augmentation by SSR504734 of the Number of Contralateral Rotations Induced by Intrastriatal Microinjection of Glycine in Mice
Glycine (0.001–1000 ng), injected by the intrastriatal route, dose-dependently and significantly (F(9,194)=5.11, P<0.001) induced contralateral rotations, with 30, 100, and 1000 ng producing significant effects (data not shown). When given in association with a subliminal dose of 0.001 ng of glycine, SSR504734 dose-dependently and significantly (F(5,66)=2.92, P<0.05) increased contralateral rotations, with a minimal efficacious dose of 1 mg/kg i.p. (Figure 5a). When tested in a separate experiment (Figure 5b), but under the same experimental conditions, SSR506204 (10 mg/kg i.p.) was inactive when compared to SSR504734 (1 mg/kg i.p.). SSR504734 was also found to induce a significant (Dunnett's post hoc test following a one-way ANOVA: F(5,93)=4.36, P<0.001) increase in contralateral rotations when microinjected alone unilaterally into the striatum (2.8±0.7 vs 8.8±1.2 vs 12.3±1.4 for vehicle, 0.001, and 0.01 pg SSR504734, respectively).

Augmentation by SSR504734 of the number of contralateral rotations induced by intrastriatal microinjection of glycine in mice. (a) Dose–effect relationship of SSR504734. (b) Lack of effects of SSR506204 as compared to SSR504734. For both panels, each bar represents the average (+SEM) number of contralateral rotations. For each group, mice were injected i.p. 30 min before an intrastriatal microinjection of glycine (0.001 ng, a subliminal dose). *P<0.05, compared to vehicle (Veh)-treated groups (Dunnett's post hoc tests following a one-way ANOVA). N=12–24 mice per group.
The noncompetitive NMDA receptor antagonist MK-801, at a dose (0.03 mg/kg i.p.) that did not significantly modify turning behavior produced by 0.001 ng glycine (5.2±1.0 vs 6.6±1.2 for vehicle and MK-801, respectively), significantly antagonized the increase (+194%) of the number of turns induced by 1 mg/kg i.p. of SSR504734 (15.3±2.1 vs 6.0±1.3, P<0.01, Dunnett's post hoc test following significant ANOVA: F(3,44)=10.39, P<0.001).
Antagonism by SSR504734 of the Decrease of [3H]acetylcholine Release Induced by the CB1 Receptor Agonist WIN55212-2 in Rat Hippocampal Slices
Addition of 0.01–1 μM WIN55212-2 to the superfusion medium caused a concentration-dependent and significant (F(3,31)=5.78, P<0.01) inhibition of electrically stimulated release of [3H]ACh (Figure 6, inset). The inhibitory effect (34.8%±3.6; Figure 6, foremost left filled square) produced by 1 μM WIN55212-2 was significantly (F(6,20)=8.39, P<0.01) counteracted by SSR504734 (Figure 6, filled squares), in a concentration-dependent manner and with an IC50=16±2 nM. When given alone, SSR504734, up to 1 μM, did not significantly affect either spontaneous (data not shown) or electrically evoked (F(3,32)=0.34, P>0.05) release of [3H]ACh (Figure 6, open squares). The enantiomer SSR506204, at 0.1 μM, failed to reverse the inhibition of [3H]ACh release induced by WIN55212-2 (Figure 6, triangle).

Antagonism by SSR504734 of the decrease of [3H]ACh release induced by the CB1 receptor agonist WIN55212-2 in rat hippocampal slices. Inset: Concentration-dependent decrease of [3H]ACh release induced by WIN55212-2. Each bar/symbol represents the mean and SEM. *P<0.05, compared to the WIN55212/Veh group (foremost left filled square; Dunnett's post hoc tests following a one-way ANOVA). N=12–16 determinations (from 3–4 independent experiments) per group, except for the absolute control and the WIN55112/Veh groups (40 determinations, from 10 independent experiments).
Antagonism by SSR504734 of the Decrease in Firing Rate of Prefrontal Cortex Neurons Induced by the CB1 Receptor Agonist WIN55212-2
Baseline firing of medial PFC cells occurred mainly in bursts of 2–4 spikes of approximately 1.5 ms duration and 0.2 mV amplitude at a spontaneous (control) frequency ranging from 0.3 to 6.0 Hz (mean±SEM: 2.5±0.3 Hz, n=20; Figure 7a, foremost left bars of the left panel). The i.v. administration of WIN55212-2 (0.1 mg/kg) elicited inhibition of firing rate in four out of the five PFC cells recorded. Inhibition was maximal (52.5±6.6%; Figure 7b, second bar from left) 5 min after administration and remained statistically significant (F(8,24)=9.69, P<0.01) for the following 20 min, before returning to baseline.

(a) Typical samples of firing rate histograms recorded from PFC neurons of the rat. Left: Inhibitory effect of WIN55212-2 (WIN, 0.1 mg/kg i.v.) when preceded by vehicle (Veh). Right: Attenuation of WIN55212-2-induced inhibition after pretreatment by SSR504734 (1 mg/kg i.v.). (b) Prevention by SSR504734, but not SSR506204, of the decrease of firing rate of PFC neurons induced by the CB1 receptor agonist WIN55212-2. All measurements were made at the time of WIN55212-induced maximum inhibition (5 min after its administration). Each bar represents the mean+SEM. *P<0.05, **P<0.01 with respect to values before administration of WIN55212-2 (Duncan's post hoc tests following a one-way ANOVA). N=4 rats per group. Note: Doses are expressed as the weight of the salt.
Administration of SSR504734 (0.3 and 1 mg/kg i.v.) 10 min prior to vehicle did not change the firing rate of PFC cells (data not shown). However, when administered prior to WIN55212-2 (0.1 mg/kg i.v.), SSR504734 dose-dependently (0.3–1 mg/kg i.v.) prevented the inhibitory effects of the CB1 agonist (Figure 7b, third and fourth bars from left). This was confirmed by the statistical analysis showing that only the SSR504734 1 mg/kg/WIN55212-2 groups were not significantly different from the absolute control group (Duncan's test following a significant one-way ANOVA: F(8,24)=3.72, P<0.01). By contrast, pretreatment with 1 mg/kg i.v. of SSR506204 (a dose that by itself was without effects on neuronal activity) was ineffective in reversing the decrease of firing induced by WIN55212-2 (F(8,24)=5.10, P<0.05; Figure, foremost right bar).
Increase by SSR504734 of Extracellular Levels of Dopamine in the Prefrontal Cortex of Freely Moving Rats
The average basal extracellular DA level in the PFC was estimated to be 99±20 pM (n=16). SSR504734 (10 and 30 mg/kg i.p.) dose-dependently (F(2,13)=7.61, P<0.01 and F(12,78)=2.99, P<0.01 for the treatment and treatment × time interaction factors, respectively) increased extracellular DA levels (maximal increase observed at 10 and 30 mg/kg i.p. at 20 min: 107±21 and 153±41% of control, respectively; Figure 8).

Increase by SSR504734 of extracellular levels of DA in the PFC of freely moving rats. Changes in DA levels are expressed as a percentage of the mean value of the four basal samples before the start of the treatment (indicated by vertical arrow). Each symbol represents the mean+SEM. **P<0.01 compared with the vehicle group at the corresponding time of sampling (Dunnett's post hoc tests following a one-way ANOVA). N=5–6 rats per group.
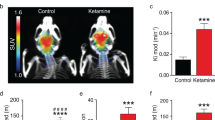
Blockade by SSR504734 of Ketamine-Induced Increase of Brain Metabolic Activity in Mice
Ketamine administration (30 mg/kg i.p.) produced robust (+38 to +70%) and significant (all F's>5.03, all P's<0.01, Newman-Keuls post hoc tests following significant ANOVAs) increases of relative 2-DG uptake in the five limbic regions studied (white bars in Figure 9). SSR504734 (10 or 30 mg/kg i.p.), by itself, did not produce significant effects on basal 2-DG uptake (+6 to +10%, and −1 to −7%, for 10 or 30 mg/kg, respectively; not reported in Figure 9). The low dose of SSR504734 partially reduced ketamine-induced increases in 2-DG uptake, and this effect was significant in the dorsal hippocampus, PFC, and cingulate cortex. At 30 mg/kg i.p., SSR504734 produced a significant and near complete reversion of the stimulatory effect of ketamine in all five brain regions studied (cross-hatched bars in Figure 9).

Blockade by SSR504734 of ketamine-induced increase of brain metabolic activity in mice. Each bar represents the average (+ SEM) uptake of 2-deoxyglucose (2-DG) for each of the five limbic structures studied. Ketamine was injected i.p. at 30 mg/kg 2 min before the 2-DG solution. **P<0.01, compared to vehicle (Veh), +P<0.05, ++P<0.01, compared to the ketamine/Veh-treated group (Newman–Keuls post hoc tests following a one-way ANOVA). N=4–6 mice per group.
Antagonism by SSR504734 of Locomotor Hyperactivity Induced by MK-801 in Mice
With respect to controls, MK-801 (0.2 mg/kg. i.p.) produced a ca three-fold increase in the number of interrupted light beams (from 618±76 to 1790±178; compare the two foremost left symbols in Figure 10a). SSR504734 dose-dependently antagonized MK-801-induced hypermotility, with the level of motility at 30 mg/kg being close to that obtained in vehicle-injected animals (curve with filled circles, Figure 10a). At this dose of 30 mg/kg that nearly completely antagonized MK-801 hyperactivity, SSR504734 was inactive by itself (foremost right square in Figure 10a), suggesting that this antagonism was not due to a nonspecific motor effect. This was confirmed by statistical analysis, with Dunnett's post hoc tests (following a one-way ANOVA: F(6,63=14.92, P<0.0001)) showing that the absolute control, the MK-801/SSR504734 (30 mg/kg), and Veh/SSR504734 (10 and 30 mg/kg) groups were significantly different from the Veh/MK-801 group.

(a) Antagonism by SSR504734 of MK-801-induced hyperactivity in mice. (b) Lack of effects of SSR506204 as compared to SSR504734 (both at 30 mg/kg i.p.). For both panels, each symbol/bar represents the average (with SEM) number of light beam interruptions recorded for 30 min, 30 min following an i.p. injection of MK-801 (MK; 0.2 mg/kg i.p.). (++)P=0.02, ++P<0.01, compared to the vehicle (Veh)/MK-801 group (Dunnett's post hoc tests following one-way ANOVAs). N=8–10 mice per group.
In a separate experiment, SSR504734, but not SSR506204 (both at 30 mg/kg i.p.), significantly antagonized MK-801-induced hyperlocomotion (Figure 10b). This was supported by the statistical analysis, with Dunnett's post hoc tests (following a one-way ANOVA: F(4,35)=11.69, P<0.0001) showing that the absolute control, the MK-801/SSR504734, and Veh/SSR506204 groups were significantly different from the Veh/MK-801 group.
Reversal by SSR504734 of MK-801-Induced Increase of the Absolute Power of the Alpha1 Band in Rat Cortex
MK-801 alone (Figure 11, white bars) induced a robust and rapid increase of the power of the alpha1 band. This increase was antagonized by SSR504734 during the first 10 min only at 10 mg/kg i.p., but totally prevented during the whole 40 min of recording at 30 mg/kg i.p. This was supported by the two-way ANOVA revealing significant effects for treatment (F(2,12)=8.72, P<0.01), time (F(5,10)=13.15, P<0.0001), and treatment × time interaction (F(10,60)=5.63, P<0.0001), and subsequent one-way ANOVAs showing significant effects of 30 mg/kg SSR504734 for each of the four times following administration of MK-801 (all F's>4.96, all P's<0.01).

Reversal by SSR504734 of MK-801-induced increase of the absolute power of the alpha1 band in rats. Each bar represents the average (+SEM) variation of the absolute power of the alpha1 frequency band expressed as a percentage of the control period. MK-801 was injected i.p. at 0.2 mg/kg. *P<0.05, **P<0.01, compared to vehicle (Veh)-treated group at each recording period (Dunnett's post hoc tests following a one-way ANOVA). N=5 rats per group.
Antagonism by SSR504734 of the Impairment of Novelty Discrimination and the Hypersensitivity to an Acute Challenge with d-Amphetamine, in Adult Rats Treated with Phencyclidine at the Neonatal Stage
Under control conditions (ie acute injection of vehicle), adult rats pretreated with vehicle at the neonatal stage (Figure 12, foremost left white bar) spent approximately four-fold more time investigating the novel rather than the familiar juvenile (NDI # 4). By contrast, adult rats neonatally pretreated with PCP presented an NDI roughly half that of vehicle-neonatal rats (compare the foremost left pair of bars in Figure 12). This indicates that PCP-neonate adult rats spent less time exploring the novel juvenile, which can be interpreted as an impairment of selective attention (see Terranova et al, in press, for in-depth discussion). Treatment with SSR504734 dose-dependently normalized this impairment: this was supported by post hoc statistical analysis (see legend of Figure 12 for details) following a two-way ANOVA with a significant neonatal pretreatment effect and acute treatment effect (F(1,8)=5.90, P<0.05 and F(3,24)=3.21, P<0.05, respectively). Note that in the vehicle-neonatal control group, SSR504734 had no effect by itself (compare the four white bars in Figure 12).

Antagonism by SSR504734 of the impairment of novelty discrimination in adult rats treated with PCP administration at the neonatal stage. Each bar represents the average (+SEM) NDI (ratio of the time spent investigating the novel juvenile divided by the time spent investigating the familiar juvenile, in seconds). Post hoc analyses following a two-way ANOVA: *P<0.05, neonatal PCP-treated compared to neonatal vehicle-treated rats, at each acute treatment condition. ##P<0.01, compared to vehicle (Veh)-treated rats, for the cohort of neonatal PCP-treated rats. N=5 rats per group.
With respect to controls, d-amphetamine produced a 151% increase in the number of interrupted infrared beams in neonatal vehicle-treated rats. This increase was more than tripled (518%) in neonatal PCP-treated rats, indicating hypersensitivity to the locomotor-enhancing effects of an acute challenge with d-amphetamine at the adult stage (compare the two foremost left pairs of bars in Figure 13a). SSR504734 dose-dependently attenuated this hypersensitivity, with a near total antagonism at 3 mg/kg i.p. This attenuation is better seen in Figure 13b, which shows the dose-dependent attenuating effect of SSR504734 on the differential sensitivity to d-amphetamine between neonatal PCP- and neonatal vehicle-treated rats. The attenuating effect of SSR504734 was confirmed by statistical analysis (see legend of Figure 13 for details) following a two-way ANOVA with a significant neonatal pretreatment effect, acute treatment effect, and neonatal × acute interaction (F(1,8)=7.06, P<0.05, F(4,32)=22.51, P<0.01, and F(4,32)=4.65, P<0.05, respectively).

(a) Antagonism by SSR504734 of the hypersensitivity to an acute challenge with d-amphetamine in adult rats treated with PCP administration at the neonatal stage. Each bar represents the average (+SEM) number of infrared beam interruptions recorded for 90 min, immediately after an injection of d-amphetamine (2 mg/kg i.p.) or vehicle, which was preceded 30 min earlier by an i.p. injection of SSR504734 or vehicle. Post hoc analyses following a two-way ANOVA: **P<0.01, neonatal PCP-treated compared to neonatal vehicle-treated rats, at each acute treatment condition. #P<0.05, ##P<0.01, compared to d-amphetamine/vehicle-injected rats (0.0 group), for the cohort of neonatal PCP-treated rats. N=5 rats per group. (b) Difference (expressed as percentage) between neonatal PCP- and vehicle-treated rats to the locomotor-enhancing effects of an acute challenge with d-amphetamine, as a function of the dose of SSR504734.
The doses of 1 and 3 mg/kg of SSR504734, which nearly completely prevented hypersensitivity to d-amphetamine in the cohort of neonatal PCP-treated rats, had by themselves no effect on spontaneous locomotor activity (recorded during the 30 min preceding administration of d-amphetamine: vehicle-neonatal group, saline: 4579.8±891.7 and SSR 1 mg/kg: 2590.4±606.3, NS; PCP-neonatal group, saline: 4911.0±605.1 and SSR 1 mg/kg: 4263.4±660.5, NS; vehicle-neonatal group, saline: 2145.8±316.2 and SSR 3 mg/kg: 3708.4±484.8, NS; PCP-neonatal group, saline: 2644.8±538.0 and SSR 3 mg/kg: 5003.8±1035.4, NS). This suggests that the effect of SSR504734 was selective against the neonatal PCP-induced hypersensitivity, and not due to a nonspecific motor effect. In addition, the basal level of activity (during the habituation period) for PCP- and vehicle-neonatal groups did not differ significantly (data not shown).
Reversion by SSR504734 of a Spontaneous Deficit of Prepulse Inhibition of the Startle Reflex in DBA/2 Mice
Under control conditions, DBA/2 mice presented a spontaneous deficit of PPI, ranging from −35±18% to 23±12%, depending on the intensity of the prepulse (see white bars in Figure 14a). By comparison, under similar experimental conditions, CD1 mice (a strain with no spontaneous deficit) showed PPI values in the range of 49±7 to 80±4% (graph not shown).

Reversion by SSR504734 of a spontaneous deficit of prepulse inhibition of the startle reflex in DBA/2 mice. (a) Prepulse inhibition expressed as a function of prepulse (PP) intensity (in dB). (b) PPI averaged across all three PP intensities. See the text for details on the calculation of PPI. For both panels, each bar represents the average+SEM. *P<0.05, **P<0.01 vs vehicle (Dunnett's post hoc tests at the corresponding prepulse intensity following a two-way ANOVA (a) or a one-way ANOVA (b)). N=8–16 mice per group. Note: Doses are expressed as the weight of the salt.
SSR504734 dose-dependently increased PPI values as compared to vehicle pretreatment, that is, reversed the spontaneous deficit of PPI presented by this strain of mice. This was confirmed by statistical analysis, with the two-way ANOVA revealing a significant treatment effect (F(2,36)=6.86, P<0.01). When PPI values were averaged across all three prepulse intensities (Figure 14b), both doses of SSR504734 were found to significantly increase PPI with respect to vehicle conditions (Dunnett's post hoc tests, following a significant one-way ANOVA: F(2,36)=6.86, P<0.01).
Basal startle amplitude (in the absence of any prepulse) was not significantly modified by SSR504734 (164±20, 202±31, and 194±38 arbitrary units, for vehicle and SSR504734 15 mg/kg and 30 mg/kg, respectively, all P's>0.05, Dunnett's post hoc tests). Note: Due to levels of basal startle that were too low for a meaningful interpretation of data, nine (out of 17) mice of the 30 mg/kg group were excluded from the PPI and basal startle amplitude analyses.
Reduction by SSR504734 of Ultrasonic Distress Vocalizations in Rat Pups
SSR504734, from 1 to 10 mg/kg s.c., dose-dependently and significantly (F(3,44)=7.21, P<0.001) decreased distress vocalizations in rat pups separated from their mother, with a near total abolition at the maximal dose tested (Figure 15a).

(a) Dose-effect reduction by SSR504734 of ultrasonic vocalization distress calls in rat pups. (b) Lack of effects of SSR506204 as compared to SSR504734. For both panels, each bar represents the mean (+SEM) number of ultrasonic distress calls emitted by pups separated from their mother for a period of 3 min. (*)P=0.05, **P<0.05, **P<0.01 vs vehicle (Dunnett's post hoc tests following a one-way ANOVA). N=8–12 pups per group.
In a separate experiment, SSR504734 and its (R,R) enantiomer SSR506204 were both administered at 10 mg/kg s.c. Post hoc analysis, following a significant treatment effect (F(2,21)=7.03, P<0.01), revealed that only the SSR504734-treated group significantly differed from the vehicle-treated control group (Figure 15b).
Improvement by SSR504734 of Physical State Degradation in Mice Subjected to a Chronic Mild Stress
SSR504734 was well tolerated after repeated administration during 4 weeks: except for the signs produced by CMS, no other overt abnormality (ie weight loss) was noted.
The CMS procedure led to a marked degradation of the physical state of the animal's coat (triangles in Figure 16), which was improved by SSR504734 (10 mg/kg i.p.; circles in Figure 16) from the first week following the beginning of treatment. This improvement remained stable over the total duration of the CMS procedure. Statistical analysis revealed that there were significant treatment, time, and treatment × time interaction factors (F(2,49)=195.13, P<0.001, F(6,294)=197.42, P<0.001, and F(12,49)=26.94, P<0.001, respectively).

Improvement by SSR504734 of the degradation of the physical state of the coat of mice during a CMS. Each symbol represents the average (+SEM) physical state score recorded during each of the 7 weeks of application of a CMS. *P<0.05, **P<0.01 compared to stressed/vehicle at each corresponding week of treatment (Newman–Keuls post hoc tests following a one-way ANOVA). N=16–19 mice per group.
Reduction by SSR504734 of Paradoxical Sleep in Rats
Over the recording period of 6 h, SSR504734 at 30 mg/kg i.p. produced significant effects on all five parameters recorded (all F's>4.45, all P's<0.05; Figure 17a). In particular, SSR504734 induced a long-lasting (5 h; data hour by hour not shown) reduction of PS: this reduction was mainly the consequence of an increased latency time to onset of the first episode (Lat PS) and, to a lesser extent, of the number of episodes of PS (Epi PS) and of a reduction of the time spent in this condition. At this dose, SSR504734 also increased the duration of W, but this effect was restricted to the first 2 h. At 10 mg/kg i.p., a slight reduction of PS, restricted to the first hour after SSR504734 administration, was the only effect observed. The (R,R) enantiomer SSR506204, at 30 mg/kg i.p., was basically devoid of activity and induced a weak reduction of PS only during the first hour after injection (data not shown).

(a) Reduction of PS by SSR504734, but not SSR506204, in rats. Each bar represents the average time spent during the drug challenge day (Day 2) as a percentage of control values obtained the day before (Day 1, control day with vehicle injection). *P<0.05, compared to values obtained on Day 1 (Dunnett's post hoc test following one-way ANOVAs on raw values for each parameter). (b) Lack of effects of SSR504734 and SSR506204 on cortical electroencephalogram activity in rats measured the day after drug challenge. Each bar represents the average time spent during the post-drug challenge day (Day 3) as a percentage of control values obtained on the control day (Day 1). W: wakefulness; SWS: slow-wave sleep; PS: paradoxical sleep; Epi PS: number of episodes of PS; Lat PS: latency time to enter PS. Cortical electroencephalogram activity was recorded over a period of 6 h. N=5–6 rats per group.
When recorded on Day 3 (24 h after drug challenge), none of the five parameters differed significantly from control values of Day 1 (Figure 17b). This shows that there was no PS rebound effect.
Table 2 reports on the in-house activity of SSR504734 in comparison with its (R,R) enantiomer SSR506204 and reference antipsychotics, in most of the in vivo tests used in this study. These summary results indicate that the different tests were validated with appropriate reference compounds, but for reasons of space, details of the results obtained with reference compounds are not discussed.
DISCUSSION
This is the first report providing a detailed account of the neurochemical, electrophysiological, and pharmacological properties of a new selective and reversible GlyT1 inhibitor, namely SSR504734. Other GlyT1 blockers have been described in the literature, but their very low affinity (sarcosine, glycyldodecylamide) or the apparent irreversible nature of their blocking effects (ALX5407) makes them less attractive tools for pharmacological exploration of GlyT1.
SSR504734 is a Selective Blocker of GlyT1 In Vitro and Ex Vivo
SSR504734 potently blocked the uptake of [14C]glycine in the native human SK-N-MC cell line expressing the GlyT1a isoform, as well as in the rat C6 cell line that contains GlyT1. This effect of SSR504734 was stereoselective since the (R,R) enantiomer SSR506204 was far less potent. SSR504734 inhibited glycine transport at human and rat GlyT1s (IC50s ca 20 nM) with a potency in between that reported for ALX5407 (3 nM, Atkinson et al, 2001; 220 nM, Herdon et al, 2001, both for hGlyT1c; 26 nM for hGlyT1b, Smith et al, 2004; 10 nM for rGlyT1a, Kinney et al, 2003) and for ORG 24598 (120 nM for hGlyT1b, Brown et al, 2001), and far above that of sarcosine or glycyldodecylamide, two earlier GlyT1 inhibitors (IC50s greater than 10 μM; present results; Javitt and Frusciante, 1997). SSR504734 was similarly potent in mice (IC50: 38±5 nM, in cortical homogenate, not reported in Materials and methods and Results), a point we verified because of the use of this species in several tests. SSR504734 displayed no in vitro activity at the GlyT2, glutamate, GABA, DA, and serotonin transporters. Unlike current antipsychotics, it had no affinity for DA receptors, and for other receptors that have been associated with various side effects, such as weight gain (5-HT2c, histaminergic H1), hypotension (alpha adrenergic), dry mouth/constipation/cognitive defects (muscarinic), sedation (histaminergic), to cite the main ones.
Ex vivo, SSR504734 blocked the uptake of glycine in mouse cortical homogenate after i.p. and p.o. administration. Analysis of the time course of ex vivo blockade of glycine uptake showed that contrary to ALX5407 (unpublished data), inhibition by SSR504734 was reversible. Reversibility with SSR504734, but not ALX5407, was also observed in vitro (potentiation of EPSCs in hippocampal slices). Reversibility of the blocking effect of GlyT1 by SSR504734 is of importance, as nonreversible blockade might constitute a potential disadvantage to the use of a compound in the clinic.
Blockade by SSR504734 of GlyT1 Augments Extracellular Levels of Glycine and Dopamine in the Prefrontal Cortex
In microdialysis experiments, the potency and efficacy of SSR504734 (10 mg/kg i.p.) to increase PFC basal level of glycine was at least comparable to that seen with ALX5407 (10 mg/kg i.p. or p.o.; unpublished data; Atkinson et al, 2001). The capacity of SSR504734 to increase extracellular levels of glycine, as well as those of DA, in the PFC of rats is expected to have several consequences. The PFC holds a privileged place among the brain structures central to the expression of schizophrenia. Evidence has accumulated to implicate hypofunctioning of the PFC in the genesis of negative symptoms and cognitive deficits (Goldman-Rakic and Selemon, 1997) that are more and more considered to constitute the core of the pathology. Deficiency of at least two neurotransmission systems has been suspected to cause hypofunctioning of the PFC: the NMDA/glutamatergic and the dopaminergic systems (Goldman-Rakic and Selemon, 1997). Augmentation by SSR504734 of prefrontal glycinergic tone is anticipated to potentiate the NMDA/glutamatergic transmission (see below) and, one step further, to re-equilibrate a deficiency of this neurotransmission. Additionally, the ability of current antipsychotics to augment prefrontal DA tone is considered to be a neurochemical marker indicative of their capacity to alleviate negative and cognitive symptoms in patients (Kapur and Remington, 1996). Together, these data suggest that SSR504734, by virtue of a bimodal neurochemical mechanism, could be effective in combating negative/cognitive deficits in schizophrenic patients. Finally, suboptimal activity of the DA system in the PFC has been suspected to lead to hyperfunctioning of the subcortical dopaminergic system, leading to the genesis of positive symptoms of schizophrenia (Grace, 1991). By virtue of its ability to potentiate DA transmission in the PFC (and hence to diminish subcortical DA tone), SSR504734 is also anticipated to possess a direct beneficial effect on positive symptoms, a hypothesis further born out by behavioral data (see below).
Augmentation by SSR504734 of Central Glycinergic Tone has a Functional Impact on Central Glutamatergic Neurotransmission
The increase in central levels of glycine induced by SSR504734 had a functional impact on central glutamatergic neurotransmission, both in vitro and in vivo. In vitro, SSR504734 potentiated NMDA-mediated eEPSCs in rat hippocampal slices. In line with these results, Wilcox and collaborators (1996) have shown that NMDA receptor-mediated eEPSCs recorded from hippocampal neurons can be markedly potentiated by glycine. There is some evidence pointing to anomalies of the hippocampus in schizophrenic patients, both at the cytoarchitectural and neurochemical levels (Heckers and Konradi, 2002). The hippocampus has been implicated in several aspects of cognitive function, among which is episodic memory, that is affected in schizophrenic patients (Rushe et al, 1999). The present data, combined with the demonstration that SSR504734 (manuscript in preparation) and the two GlyT1 inhibitors NFPS (Kinney et al, 2003) and CP 802,079 (Martina et al, 2004) enhanced long term potentiation in the hippocampus in vivo, suggest a beneficial effect of GlyT1 inhibitors on hippocampal-dependent forms of memory deficient in schizophrenia. Furthermore, in anesthetized rats, SSR504734 enhanced a DA voltametric signal in the nucleus accumbens, elicited by electrical stimulation of the amygdala. Since this DA signal was shown to be NMDA receptor-dependent, this enhancement provides a supplementary argument for a potentiation of the NMDA neurotransmission consecutive to blockade of GlyT1 by SSR504734 (Leonetti et al, submitted).
Systemic administration of SSR504734 dose-dependently increased the number of contralateral rotations in mice induced by intrastriatal injection of a subliminal dose of glycine, and SSR504734 was also found to induce contralateral rotations when microinjected alone unilaterally into the striatum. The effect of SSR504734 was antagonized by MK-801, at a dose that had no effect by itself, further suggesting that the enhancement of behavioral output (increased number of rotations) was subsequent to a potentiation of the glutamatergic/NMDA neurotransmission.
Together, these in vitro and in vivo results strengthen the suggestion (Bergeron et al, 1998; Chen et al, 2003) that glycine concentration in the vicinity of NMDA receptors is kept at subsaturating levels by GlyT1, and that increasing glycine concentration by blockade of this transporter results in enhanced NMDA-mediated function.
SSR504734 Normalizes Hypofunctioning of the NMDA/Glutamatergic system in Two Brain Structures Thought to be Implicated in Schizophrenia
Among the brain structures that have been suspected to be linked to the etiology of schizophrenia, two of them hold a prominent place: the hippocampus and the PFC. The former has been repeatedly shown to present subtle neuroanatomical anomalies in patients (Heckers and Konradi, 2002), while hypofunctioning of the latter is thought to play a role in the genesis of negative symptoms (Goff and Evins, 1998), and both are implicated in the control of cognitive functions (in particular, memory and attentional processes) that are affected in the pathology. Excitatory synaptic transmission is negatively modulated by presynaptic CB1 receptors, presumably through blockade of neurotransmitter release and in particular of glutamate (for review see Schlicker and Kathmann, 2001). CB1 receptor agonists such as WIN55212-2 have been shown to inhibit [3H]ACh efflux in rat hippocampal slices (Gifford and Ashby, 1996). The present study showed that SSR504734 nearly completely reversed the decrease in [3H]ACh efflux mediated by WIN55212-2 in a similar type of hippocampal in vitro preparation. An equivalent reversion was obtained with 10−5 M NMDA or 10−4 M glycine (data not shown), strongly suggesting that effects of SSR504734 are linked to activation of NMDA receptors. Considering the importance of the cholinergic system in memory processes (Gold, 2003), the demonstration that SSR504734 can re-establish normal levels of central cholinergic transmission predicts a potential activity of the compound against memory disturbances, not only in schizophrenia, but also possibly in other pathologies such as Alzheimer's disease. All in all, these data complement those obtained in the eEPSCs experiment (see above), and provide robust experimental arguments that SSR504734 can have a facilitatory or normalizing effect on basal or reduced hippocampal neuronal activity, respectively.
In anesthetized rats, the diminished spontaneous firing rate of PFC neurons induced by WIN55212-2 was fully reversed by administration of SSR504734. Normal functioning of the PFC closely depends on glutamatergic neurotransmission, mainly via the glutamate/NMDA system, and disturbance of this system has been implicated in schizophrenia (see for review Konradi and Heckers, 2003). In vitro, CB1 receptor agonists have been shown to depress glutamatergic synaptic transmission in various brain regions including the PFC (Auclair et al, 2000). Normalization by SSR504734 of reduced PFC neurons activity provides further evidence that enhancement of glycinergic tone by blockade of GlyT1 can reverse hypofunctioning (at least on electrophysiological parameters) of this structure. It can thus be anticipated that such a compound could re-establish normal neuronal activity in a brain structure (the PFC) suspected to be hypoactive in schizophrenic patients. It should be stressed that the possibility that the beneficial effects of SSR504734 in these two experiments ([3H]ACh hippocampal efflux and PFC spontaneous firing rate) stem from a direct antagonist activity of SSR504734 on CB1 receptors can be excluded, since the compound has no demonstrated affinity for this receptor (IC50>1 μM).
SSR504734 Shows Activity in Various Tests Predictive of Antipsychotic Activity
Evidence for a potential antipsychotic activity of SSR504734 was obtained in various neurochemical and behavioral tests considered to detect activity against positive and/or negative symptoms of schizophrenia.
Augmented 2-DG uptake is thought to reflect an increase in the metabolic rate, that is, in the level of neuronal activity. SSR504734 normalized this relative increase of 2-DG uptake in the PFC and in other subcortical limbic regions induced by an acute subanesthetic dose of ketamine in mice. Similar patterns of increased 2-DG uptake with ketamine have been reported in the literature (Duncan et al, 1998a, 1998b; Miyamoto et al, 2000). These increases could be completely blocked by acute administration of atypical antipsychotic drugs such as clozapine, but not by typical compounds such as haloperidol (Duncan et al, 1998b). In the clinic, ketamine-induced psychotic symptoms in volunteers correlate positively with increased metabolic activity in the frontal cortex (Breier et al, 1997; Vollenweider et al, 1997). The reduction by SSR504734 of an augmented cortical metabolic activity in mice (that resulted from a hypoglutamatergic state) is thus indicative of a potential activity of the compound against florid symptomatology.
At the behavioral level, SSR504734 antagonized MK-801-induced hyperactivity in mice and the increase of spectral energy in the cortical alpha1 band in rats. Harsing and colleagues (2003) reported similar effects of NFPS and/or ORG 24461 for PCP-induced hyperactivity in mice and changes in EEG spectral power in rats. MK-801-induced hyperactivity and modification of EEG pattern are, in our hands, sensitive to haloperidol and clozapine, possibly suggesting that activity in these tests is predictive of efficacy only against positive symptoms, since haloperidol is considered as poorly efficacious against negative and cognitive deficits.
DBA/2 mice, as previously reported (Olivier et al, 2001; Kinney et al, 2003), present a low basal level of PPI of the startle reflex. In other words, they show a spontaneous deficit of PPI. SSR504734 dose-dependently enhanced levels of PPI, that is, reduced this innate PPI deficit. A similar reversing effect was reported in DBA/2 mice following treatment with clozapine, haloperidol, and risperidone (McCaughran et al, 1997; Olivier et al, 2001), and more recently with (+)NFPS, the more active enantiomeric form of NFPS (Kinney et al, 2003). Additionally, glycine and ORG 24598 (the active R(−) enantiomer of ORG 24461) were also found to reverse a PPI deficit in adult rats that have undergone a ventral hippocampal lesion at the neonatal stage (Le Pen et al, 2003). All these data underline the potential of proglycinergic strategies to reverse deficits of PPI of different nature. Schizophrenic patients are known to present a deficit of PPI (Braff et al, 1978), thought to be an overt manifestation of an underlying abnormal ability to process information. The effects of SSR504734 indicate that the compound could alleviate this abnormality, considered as a pivotal element in the expression of the pathology.
The neonatal PCP model is based on the neurodevelopmental concept of schizophrenia (Weinberger, 1986; Lieberman et al, 1997). Wang and colleagues (2001, 2003) have shown that administration of high doses of PCP to rat pups produced long-term behavioral changes, associated with neuronal alterations, at the adolescent or adult stage. SSR504734 was found to have beneficial effects in adult rats neonatally treated with PCP on two behavioral aspects relevant to the pathology: (1) hypersensitivity to an acute challenge with d-amphetamine (that has been reported in schizophrenic patients during acute psychotic episodes; Laruelle, 2000) and (2) selective attention deficit (a cognitive process that is greatly affected in schizophrenia, and considered to be a predominant characteristic of the disease; Brébion et al, 2000). The beneficial effects of SSR504734 on the former are consonant with the recent findings that chronic treatment with NFPS or glycine prevented the potentiation of d-amphetamine-induced DA release in the striatum seen with chronic administration of PCP (Javitt et al, 2004). To the extent that this hypersensitivity to d-amphetamine reflects an abnormal sensitivity of subcortical DA systems, conducive to the genesis of florid symptoms in schizophrenic patients (Laruelle, 2000), the blunting effect of SSR504734 provides an additional experimental argument for a potential activity of the compound against positive symptomatology. This normalization of impairment of selective attention in rats and of spontaneous deficit of PPI (a putative marker of information processing) in DBA/2 mice is promising in terms of a beneficial activity of SSR504734 on those cognitive processes that are impaired in schizophrenic patients. Furthermore, the recent demonstration that the direct glycine agonist d-serine reversed the deleterious effects produced by a similar neonatal PCP treatment in a spatial memory task in rats (Andersen and Pouzet, 2004) buttresses the notion that a proglycinergic strategy might have a positive impact on multiple facets of cognitive deficiency in schizophrenia. This particular aspect deserves further investigation.
SSR504734 has Additional Antidepressant/Anxiolytic-Like Activity
SSR504734 was active in tests predictive of antidepressant and/or anxiolytic activity. Administered acutely, it reduced ultrasonic distress calls in pups separated from their mother, as observed with the antidepressant fluoxetine and anxiolytics such as benzodiazepines and 5-HT1A receptor agonists (Gardner, 1985; Fish et al, 2000). SSR504734 also increased the latency time to enter the paradoxical stage of sleep, an effect typically observed with antidepressants such as fluoxetine and imipramine (Slater et al, 1978; Kleinlogel, 1982). In addition, given chronically, it prevented physical degradation of mice subjected to a CMS, an effect shared with other established or putative antidepressant/anxiolytic compounds such as fluoxetine or the corticotrophin-releasing factor (1) receptor antagonist SSR125543 (Griebel et al, 2002). All in all, these pharmacological data concord with those of a very recent preliminary report mentioning a reduction in anxiety level in GlyT1 heterozygous knockout mice (Vanhoof et al, 2004).
Evidence for an antidepressant/anxiolytic potential of SSR504734 might seem rather dissonant with other preclinical data showing that antiglutamatergic compounds such as PCP and MK-801 (noncompetitive antagonists), or CCP, CPPene, and CGS 19755 (competitive antagonists) have efficacy in some animal models of anxiety (see Bergink et al, 2004 for recent review). However, not only the confirmation of the anxiolytic potential of these antiglutamatergic compounds does not seem to have been established at the clinical level, but there are even indications that these compounds might possess anxiogenic properties. An open trial with MK-801 in subjects with anxiety disorder was discontinued due to exacerbation of symptoms in the majority of patients (Reimherr et al, 1986). Furthermore, CPPene has been reported to produce restlessness, and CGS 19755 (selfotel) light-headedness, anxiety, agitation, and confusion (see review by Muir and Lees, 1995). In the light of these clinical considerations, the possibility that a compound that potentiates the NMDA/glutamatergic transmission (such as SSR504734) might have anxiolytic/antidepressant activity is not so incongruous. A more in-depth investigation of the profile of SSR504734 in other models predictive of antidepressant and/or anxiolytic activity is currently under way. In any case, anxiety and depressive states are two comorbid elements that are commonly observed in schizophrenic patients, and considered as key elements of the interictal phases of the pathology, so that aggressive management of these elements could delay and/or attenuate the severity of subsequent episodes, and lead to better stabilization of the condition.
Safety Profile of SSR504734
SSR504734 does not produce catalepsy up to 30 mg/kg i.p. in mice (data not shown), indicating that it should not induce extrapyramidal signs in patients; in this regard, it conforms to current atypical antipsychotics such as clozapine, amisulpride, olanzapine, and quietiapine. Additionally, it does not increase levels of prolactin in rats (up to 30 mg/kg i.p.; data not shown), a side effect seen with most antipsychotics.
Conclusion
SSR504734 is a potent, selective, and orally active GlyT1 inhibitor, exhibiting activity in animal models of schizophrenia and anxiety/depression. Its activity was found to be enantioselective in numerous in vitro, ex vivo, and in vivo tests, since its (R,R) enantiomer SSR506204 was less active or devoid of effects. Its mechanism of action targets what is considered as being one of the primary causes of schizophrenia, namely a hypoglutamatergic state. As such, it is expected to be efficacious not only against positive but also negative symptoms and cognitive deficits, as well as comorbid depression and anxiety states. The potential use of GlyT1 inhibitors as new therapeutic approaches for the treatment of schizophrenia is further reinforced by the very recent findings of a double-blind placebo-controlled study showing that the weak GlyT1 inhibitor sarcosine, given in augmentation with risperidone, significantly reduced positive, negative, and cognitive symptoms, with improvements comparable to those previously seen with the full glycine site agonist d-serine (Tsai et al, 2004).
References
Allen RM, Young SJ (1978). Phencyclidine-induced psychosis. Am J Psychiatry 135: 1081–1084.
Andersen JD, Pouzet B (2004). Spatial memory deficits induced by perinatal treatment of rats with PCP and reversal effect of D-serine. Neuropsychopharmacology 29: 1080–1090.
Atkinson BN, Bell SC, De Vivo M, Kowalski LR, Lechner SM, Ognyanov VI et al (2001). ALX 5407: a potent, selective inhibitor of the hGlyT1 glycine transporter. Mol Pharmacol 60: 1414–1420.
Aubrey KR, Vandenberg RJ (2001). N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy) propyl]sarcosine (NFPS) is a selective persistent inhibitor of glycine transport. Br J Pharmacol 134: 1429–1436.
Auclair N, Otani S, Soubrie P, Crepel F (2000). Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J Neurophysiol 83: 3287–3293.
Bergeron R, Meyer TM, Coyle JT, Greene RW (1998). Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci USA 95: 15730–15734.
Bergink V, van Megen HJGM, Westenberg HGM (2004). Glutamate and anxiety. Eur Neuropsychopharmacol 14: 175–183.
Bert L, Robert F, Denoroy L, Stoppini L, Renaud B (1996). Enhanced temporal resolution for the microdialysis monitoring of catecholamines and excitatory amino acids using capillary electrophoresis with laser-induced fluorescence detection. Analytical developments and in vitro validations. J Chromatogr A 755: 99–111.
Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L (1978). Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15: 339–343.
Brébion G, Smith MJ, Gorman JM, Malaspina D, Sharif Z, Amador X (2000). Memory and schizophrenia: differential link of processing speed and selective attention with two levels of encoding. J Psychiatr Res 34: 121–127.
Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D (1997). Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry 154: 805–811.
Brown A, Carlyle I, Clark J, Hamilton W, Gibson S, McGarry G et al (2001). Discovery and SAR of Org 24598—a selective glycine uptake inhibitor. Bioorg Med Chem Lett 11: 2007–2009.
Carlsson A, Lindqvist M (1963). Effects of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol 20: 140–144.
Chen L, Muhlhauser M, Yang CR (2003). Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol 89: 691–703.
Cubelos B, Gimenez C, Zafra F (2005). Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb Cortex 15: 448–459.
Curet O, Damoiseau G, Aubin N, Sontag N, Rovei V, Jarreau FX (1996). Befloxatone, a new reversible and selective monoamine oxidase-A inhibitor. I. Biochemical profile. J Pharmacol Exp Ther 277: 253–264.
Dargazanli G, Estenne-Bouhtou G, Magat P, Marabout B, Medaisko F, Roger P et al (2003). Preparation of N-[phenyl(piperidin-2-yl)methyl]benzamides as specific inhibitors of glycine transporters GlyT1 and/or GlyT2. Patent WO2003089411-A1.
Duncan GE, Leipzig JN, Mailman RB, Lieberman JA (1998b). Differential effects of clozapine and haloperidol on ketamine-induced brain metabolic activation. Brain Res 812: 65–75.
Duncan GE, Moy SS, Knapp DJ, Mueller RA, Breese GR (1998a). Metabolic mapping of the rat after subanesthetic doses of ketamine: potential relevance to schizophrenia. Brain Res 787: 181–190.
Evins AE, Amico E, Posever TA, Toker R, Goff DC (2002). D-Cycloserine added to risperidone in patients with primary negative symptoms of schizophrenia. Schizophr Res 56: 19–23.
Fish EW, Sekinda M, Ferrari PF, Dirks A, Miczek KA (2000). Distress vocalizations in maternally separated mouse pups: modulation via 5-HT(1A), 5-HT(1B) and GABA(A) receptors. Psychopharmacology 149: 277–285.
Franklin KBJ, Paxinos G (1997). The Mouse Brain in Stereotaxic Coordinates. Academic Press: San Diego.
Gardner CR (1985). Distress vocalization in rat pups. A simple screening method for anxiolytic drugs. J Pharmacol Methods 14: 181–187.
Gifford AN, Ashby CR (1996). Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55212-2 and is potentiated by the cannabinoid antagonist, SR141716A. J Pharmacol Exp Ther 277: 1431–1436.
Goff DC, Evins AE (1998). Negative symptoms in schizophrenia: neurobiological models and treatment response. Harv Rev Psychiatry 6: 59–77.
Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA et al (1999). A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry 56: 21–27.
Goff DC, Tsai G, Manoach DS, Coyle JT (1995). Dose-finding trial of D-cycloserine added to neuroleptics for negative symptoms in schizophrenia. Am J Psychiatry 152: 1213–1215.
Gold PE (2003). Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol Learn Mem 80: 194–210.
Goldman-Rakic PS, Selemon LD (1997). Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull 23: 437–458.
Gomeza J, Ohno K, Betz H (2003). Glycine transporter isoforms in the mammalian central nervous system: structures, functions and therapeutic promises. Curr Opin Drug Discov Dev 5: 675–682.
Gomeza J, Zafra F, Olivares L, Gimenez C, Aragon C (1995). Regulation by phorbol esters of the glycine transporter (GLYT1) in glioblastoma cells. Biochim Biophys Acta 1233: 41–46.
Grace AA (1991). Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 41: 1–24.
Griebel G, Simiand J, Steinberg R, Jung M, Gully D, Roger P et al (2002). 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1, 3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotrophin-releasing factor(1) receptor antagonist. II. Characterization in rodent models of stress-related disorders. J Pharmacol Exp Ther 301: 333–345.
Harsing Jr LG, Gacsalyi I, Szabo G, Schmidt E, Sziray N, Sebban C et al (2003). The glycine transporter-1 inhibitors NFPS and Org 24461: a pharmacological study. Pharmacol Biochem Behav 74: 811–825.
Hashimoto A, Oka T (1997). Free D-aspartate and D-serine in the mammalian brain and periphery. Prog Neurobiol 52: 325–353.
Heckers S, Konradi C (2002). Hippocampal neurons in schizophrenia. J Neural Transm 109: 891–905.
Herdon HJ, Godfrey FM, Brown AM, Coulton S, Evans JR, Cairns WJ (2001). Pharmacological assessment of the role of the glycine transporter Glyt-1 in mediating high-affinity glycine uptake by rat cerebral cortex and cerebellum synaptosomes. Neuropharmacology 10: 88–96.
Heresco-Levy U, Ermilov M, Shimoni J, Shapira B, Silipo G, Javitt DC (2002). Placebo-controlled trial of D-cycloserine added to conventional neuroleptics, olanzapine, or risperidone in schizophrenia. Am J Psychiatry 159: 480–482.
Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Silipo G, Lichtenstein M (1999). Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psychiatry 56: 29–36.
Heresco-Levy U, Javitt DC, Ermilov M, Silipo G, Shimoni J (1998). Double-blind, placebo-controlled, crossover trial of D-cycloserine adjuvant therapy for treatment-resistant schizophrenia. Int J Neuropsychopharmacol 1: 131–135.
Javitt DC, Balla A, Burch S, Suckow R, Xie S, Sershen H (2004). Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology 29: 300–307.
Javitt DC, Frusciante M (1997). Glycyldodecylamide, a phencyclidine behavioral antagonist, blocks cortical glycine uptake: implications for schizophrenia and substance abuse. Psychopharmacology 129: 96–98.
Javitt DC, Silipo G, Cienfuegos A, Shelley AM, Bark N, Park M et al (2001). Adjunctive high-dose glycine in the treatment of schizophrenia. Int J Neuropsychopharmacol 4: 385–391.
Javitt DC, Zukin SR (1991). Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308.
Johnson JW, Ascher P (1987). Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325: 529–531.
Kapur S, Remington G (1996). Serotonin–dopamine interaction and its relevance to schizophrenia. Am J Psychiatry 153: 466–476.
Kim KM, Kingsmore SF, Han H, Yang-Feng TL, Godinot N, Seldin MF et al (1994). Cloning of the human glycine transporter type 1: molecular and pharmacological characterization of novel isoform variants and chromosomal localization of the gene in the human and mouse genomes. Mol Pharmacol 45: 608–617.
Kinney GG, Sur C, Burno M, Mallorga PJ, Williams JB, Figueroa DJ et al (2003). The glycine transporter type 1 inhibitor N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine potentiates NMDA receptor-mediated responses in vivo and produces an antipsychotic profile in rodent behavior. J Neurosi 23: 7586–7591.
Kleinlogel H (1982). The rat paradoxical sleep as a pharmacological test model. In: Herrmann WM (ed). EEG in Drug Research. Gustave Fisher: Stuttgart. pp 75–88.
Konradi C, Heckers S (2003). Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther 97: 153–179.
Kopp C, Vogel E, Rettori MC, Delagrange P, Misslin R (1999). The effects of melatonin on the behavioural disturbances induced by chronic mild stress in C3H/He mice. Behav Pharmacol 10: 73–83.
Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995). Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13: 9–19.
Laruelle M (2000). The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res Brain Res Rev 31: 371–384.
Le Pen G, Kew J, Alberati D, Borroni E, Heitz MP, Moreau JL (2003). Prepulse inhibition deficits of the startle reflex in neonatal ventral hippocampal-lesioned rats: reversal by glycine and a glycine transporter inhibitor. Biol Psychiatry 54: 1162–1170.
Lewis R (2004). Should cognitive deficit be a diagnostic criterion for schizophrenia? J Psychiatry Neurosci 29: 102–113.
Lieberman JA, Sheitman BB, Kinon BJ (1997). Neurochemical sensitization in the pathophysiology of schizophrenia: deficits and dysfunction in neuronal regulation and plasticity. Neuropsychopharmacology 17: 205–229.
Mallorga PJ, Williams JB, Jacobson M, Marques R, Chaudhary A, Conn PJ et al (2003). Pharmacology and expression analysis of glycine transporter GlyT1 with [3H]-(N-[3-4′fluorophenyl)-3-(4′phenylphenoxy) propyl]) sarcosine. Neuropharmacology 45: 585–593.
Martina M, Gorfinkel Y, Halman S, Lowe JA, Periyalwar P, Schmidt CJ et al (2004). Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol 557: 489–500.
McCaughran Jr J, Mahjubi E, Decena E, Hitzemann R (1997). Genetics, haloperidol-induced catalepsy and haloperidol-induced changes in acoustic startle and prepulse inhibition. Psychopharmacology 134: 131–139.
Millan MJ (2002). N-methyl-D-aspartate receptor-coupled glycineB receptors in the pathogenesis and treatment of schizophrenia: a critical review. Curr Drug Targets CNS Neurol Disord 1: 191–213.
Miyamoto S, Leipzig JN, Lieberman JA, Duncan GE (2000). Effects of ketamine, MK-801 and amphetamine on regional brain 2-deoxyglucose uptake in freely moving mice. Neuropharmacology 22: 400–412.
Muir KW, Lees KR (1995). Clinical experience with excitatory amino acid antagonist drugs. Stroke 26: 503–513.
Olivier B, Leahy C, Mullen T, Paylor R, Groppi VE, Sarnyai Z et al (2001). The DBA/2J strain and prepulse inhibition of startle: a model system to test antipsychotics? Psychopharmacology 156: 284–290.
Olney JW, Farber NB (1995). Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 52: 998–1007.
Paxinos G, Watson C (1998). The Rat Brain in Stereotaxic Coordinates. Academic Press: Sydney.
Poncelet M, Santucci V, Paul R, Gueudet C, Lavastre S, Guitard J et al (1993). Neuropharmacological profile of a novel and selective ligand of the sigma site: SR31742. Neuropharmacology 6: 605–615.
Reimherr FW, Wood DR, Wender PH (1986). The use of MK 801, a novel sympathomimetic, in adults with attention deficit disorder, residual type. Psychopharmacol Bull 22: 237–242.
Riedel G, Platt B, Micheau J (2003). Glutamate receptor function in learning and memory. Behav Brain Res 140: 1–47.
Rushe TM, Woodruff PW, Murray RM, Morris RG (1999). Episodic memory and learning in patients with chronic schizophrenia. Schizophr Res 35: 85–96.
Schlicker E, Kathmann M (2001). Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 22: 565–572.
Seeman P, Lee T, Chau-Wong M, Wong K (1976). Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 261: 717–719.
Slassi A, Egle I (2004). Recent progress in the use of glycine transporter-1 inhibitors for the treatment of central and peripheral nervous system diseases. Expert Opin Ther Patents 12: 201–214.
Slater IH, Jones GT, Moore RA (1978). Inhibition of REM sleep by fluoxetine, a specific inhibitor of serotonin uptake. Neuropharmacology 17: 383–389.
Smith G, Ruhland T, Mikkelsen G, Andersen K, Christoffersen CT, Alifrangis LH et al (2004). The synthesis and SAR of 2-arylsulfanyl-phenyl piperazinyl acetic acids as glyT-1 inhibitors. Bioorg Med Chem Lett 14: 4027–4030.
Tamminga CA, Holcomb HH, Gao XM, Lahti AC (1995). Glutamate pharmacology and the treatment of schizophrenia: current status and future directions. Int Clin Psychopharmacol 10(Suppl 3): 29–37.
Terranova JP, Chabot C, Barnouin M-C, Perrault G, Depoortere R, Griebel G et al. SSR181507, a dopamine D2 receptor antagonist and 5-HT1A receptor agonist, alleviates disturbances of novelty discrimination in rats, a model of selective attention deficit. Psychopharmacology (in press).
Tsai G, Lane H, Young PJ, Lane N, Chong M (2004). Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 55: 452–456.
Tsai G, Yang P, Chung LC, Lange N, Coyle JT (1998). D-Serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 44: 1081–1089.
Vandenberg RJ, Aubrey KR (2001). Glycine transport inhibitors as potential antipsychotic drugs. Expert Opin Ther Targets 5: 507–518.
Vanhoof G, Wetsel WC, Rodriguiz RM, Eldridge M, Berben M, Moechars D et al (2004). Glycine transporter 1 heterozygotes display reduced anxiety in novel environments. Program No. 504.13. 2004 Abstract Viewer/Itinerary Planner Washington, DC: Society for Neuroscience, Online.
Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J et al (1997). Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol 7: 9–24.
Wang C, McInnis J, Ross-Sanchez M, Shinnick-Gallagher P, Wiley JL, Johnson KM (2001). Long-term behavioral and neurodegenerative effects of perinatal phencyclidine administration: implications for schizophrenia. Neuroscience 107: 535–550.
Wang C, McInnis J, West JB, Bao J, Anastasio N, Guidry JA et al (2003). Blockade of phencyclidine-induced cortical apoptosis and deficits in prepulse inhibition by M40403, a superoxide dismutase mimetic. J Pharmacol Exp Ther 304: 266–272.
Weinberger DR (1986). The pathogenesis of schizophrenia: a neurodevelopmental theory. In: Nasrallah HAW, Weinberger DR (eds). The Neurology of Schizophrenia. Elsevier: Amsterdam. pp 397–406.
Wilcox KS, Fitzsimonds RM, Johnson B, Dichter MA (1996). Glycine regulation of synaptic NMDA receptors in hippocampal neurons. J Neurophysiol 76: 3415–3424.
Willner P, Muscat R, Papp M (1992). Chronic mild stress-induced anhedonia: a realistic animal model of depression. Neurosci Biobehav Rev 16: 525–534.
Acknowledgements
We thank M Bensaid for the characterization of the subtype of GlyT1 in SK-N-MC cells, and we express our gratitude to the following persons for their expert technical assistance: C Aliaga, M-C Barnouin, Y Claustre, J Guitard, M L'Hermitte, MT Lucas, P Magat, C Marc, F Medaisko, AM Poisson, and C Veronique. We are also grateful to A-M Thierry, J-M Deniau, and Y Gioanni, from INSERM U 114, Collège de France, Paris, for their helpful assistance and advices on the in vivo electrophysiological studies. Finally, we are indebted to John Alexander for his help with the use of the English language.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Depoortère, R., Dargazanli, G., Estenne-Bouhtou, G. et al. Neurochemical, Electrophysiological and Pharmacological Profiles of the Selective Inhibitor of the Glycine Transporter-1 SSR504734, a Potential New Type of Antipsychotic. Neuropsychopharmacol 30, 1963–1985 (2005). https://doi.org/10.1038/sj.npp.1300772
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300772
Keywords
This article is cited by
-
Novel Proline Transporter Inhibitor (LQFM215) Presents Antipsychotic Effect in Ketamine Model of Schizophrenia
Neurochemical Research (2024)
-
The phosphodiesterase-4 and glycine transporter-1 inhibitors enhance in vivo hippocampal theta network connectivity and synaptic plasticity, whereas D-serine does not
Translational Psychiatry (2020)
-
Comparative Pro-cognitive and Neurochemical Profiles of Glycine Modulatory Site Agonists and Glycine Reuptake Inhibitors in the Rat: Potential Relevance to Cognitive Dysfunction and Its Management
Molecular Neurobiology (2020)
-
Assessment of a glycine uptake inhibitor in animal models of effort-related choice behavior: implications for motivational dysfunctions
Psychopharmacology (2017)
-
Selective Mitochondrial Targeting Exerts Anxiolytic Effects In Vivo
Neuropsychopharmacology (2016)
