
Abstract
Several lines of evidence suggest that oxidative stress might contribute to neurotoxicity in the dopaminergic nerve terminals after administration of methamphetamine (MAP). We undertook the present study to determine whether intravenous administration of N-acetyl-L-cysteine (NAC), a potent antioxidant drug, could attenuate the reduction of dopamine transporter (DAT) in the striatum of monkey brain after administration of MAP. Positron emission tomography studies demonstrated that repeated administration of MAP (2 mg/kg as a salt, four times at 2-h intervals) significantly decreased the accumulation of radioactivity in the striatum after intravenous administration of [11C]β-CFT. In contrast, the binding of [11C]SCH 23390 to dopamine D1 receptors in the monkey striatum was not altered after the administration of MAP. A bolus injection of NAC (150 mg/kg, i.v.) 30 min before MAP administration and a subsequent continuous infusion of NAC (12 mg/kg/h, i.v.) over 8.5 h significantly attenuated the reduction of DAT in the monkey striatum 3 weeks after the administration of MAP. These results suggest that NAC could attenuate the reduction of DAT in the monkey striatum after repeated administration of MAP. Therefore, it is likely that NAC would be a suitable drug for treatment of neurotoxicity in dopaminergic nerve terminals related to chronic use of MAP in humans.
Similar content being viewed by others


INTRODUCTION
The abuse of methamphetamine (MAP), a potent psychostimulant, is an extremely serious and growing problem in the world. The action of MAP is thought to involve rapid entry into the brain, followed by influx into monoaminergic terminals, interaction with vesicular monoaminergic transporter, entry into monoaminergic vesicles and displacement of monoamines into the cytoplasm of the terminals, and subsequent release of the monoamines into the synaptic cleft (Cadet et al, 2003). Recent studies using positron emission tomography (PET) suggest that chronic use of MAP causes the reduction of dopamine transporter (DAT) in the human brain (McCann et al, 1998; Sekine et al, 2001, 2003; Volkow et al, 2001a, 2001b), suggesting the neurotoxic effects of MAP in the human brain. These findings are supported by a report demonstrating that the densities of DAT are significantly decreased in the postmortem brain striatum of chronic MAP users (Wilson et al, 1996). However, the precise mechanisms underlying MAP-induced neurotoxicity in dopaminergic nerve terminals of the brain are currently not known (Davidson et al, 2001; Cadet et al, 2003).
Oxidative stress generated by an imbalance between reactive oxygen species (ROS; hydrogen peroxide, superoxide radical, and hydroxyl radical) and antioxidants might contribute to the neurotoxicity of MAP in the brain (Imam et al, 2001; Davidson et al, 2001; Cadet et al, 2003). N-acetyl-L-cysteine (NAC), the acetylated variant of the amino acid L-cysteine, is an excellent source of sulfhydryl (SH) groups and is converted in the body into metabolites capable of stimulating glutathione synthesis, promoting detoxification, and acting directly as free radical scavengers (Kelly, 1998). In addition to its mucolytic action, NAC has been studied and utilized to treat conditions characterized by decreased glutathione or oxidative stress such as HIV infection, cancer, and heart disease (Kelly, 1998). Owing to its known characteristics, NAC has been used as a tool for investigating the role of ROS in numerous biological and pathological processes (Kelly, 1998; Zafarullah et al, 2003). Recently, we reported that NAC significantly attenuated 6-hydroxydopamine-induced apoptotic neuronal cell death in human neuroblastoma SK-N-SH cells, suggesting that NAC could work as a beneficial dopaminergic neuron-survival factor (Shimizu et al, 2002). In addition, NAC directly modifies the activity of several proteins by its reducing activity (Zafarullah et al, 2003). Based on the role of oxidative stress in the neurotoxicity of MAP in the brain (Imam et al, 2001; Davidson et al, 2001; Cadet et al, 2003), it is interesting to study the effects of the antioxidant NAC on MAP-induced neurotoxicity in the brain.
We reported recently that pretreatment with NAC significantly attenuates hyperlocomotion, development of sensitization, and neurotoxicity after the administration of MAP in rats, suggesting that NAC might be a suitable drug for treatment of MAP abuse (Fukami et al, 2004). The purpose of the present study was to discover therapeutic drugs to prevent or protect against neurotoxicity in dopaminergic terminals in chronic MAP abusers. We performed the present PET study to determine whether intravenous administration of NAC could attenuate the reduction of DAT in the striatum of monkey brain after administration of MAP. To minimize the effects of anesthetics on the behavior of a labeled compound in vivo, we performed PET scans of monkeys in the conscious state (Onoe et al, 1994; Tsukada et al, 1999, 2000, 2001, 2002).
MATERIALS AND METHODS
Subjects
Six young-adult male rhesus monkeys (Macaca mulatta) weighing from 4 to 6 kg were used for PET measurements. Monkeys were maintained and handled in accordance with the recommendations of the US National Institutes of Health and also the guidelines of the Central Research Laboratory, Hamamatsu Photonics (Hamakita, Shizuoka, Japan). Over the course of 3 months, the monkeys were trained to sit on a chair twice a week. The magnetic resonance images (MRIs) of all monkeys were obtained with a Toshiba MRT-50A/II (0.5 T) under anesthesia with pentobarbital. The stereotactic coordinates of PET and MRI were adjusted based on the orbitomeatal (OM) line with monkeys secured in a specially designed head holder (Takechi et al, 1994). At least 1 month before the PET study, an acrylic plate, with which the monkey was fixed to the monkey chair, was attached to the head under pentobarbital anesthesia as described previously (Onoe et al, 1994).
Drug Administration
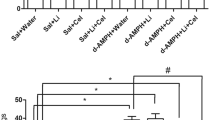
MAP hydrochloride (Dainippon Pharmaceuticals Ltd, Osaka, Japan, 2 mg/kg as a salt, four times at 2-h intervals) was administered intramuscularly into each monkey (Figure 1). We used that dose because it is reported to produce long-term neurotoxic effects on the brain of baboons (Villemagne et al, 1998). Also, such a dose regimen closely approximates the binge use of MAP by some humans (20–40 mg every 2–3 h) (Konuma, 1994). For administration of NAC, subjects received a bolus of NAC (Wako Pure Chemicals Ltd, Tokyo, Japan, 150 mg/kg, i.v.) 30 min before administration of MAP and a subsequent continuous infusion of NAC (12 mg/kg/h, i.v.) over 8.5 h, with a slight modification of the method reported previously (Molnar et al, 1999; Rank et al, 2000) (Figure 1).

Schedule of treatment of MAP and/or NAC in monkeys.
Synthesis of [11C][-]Labeled Compounds
Carbon-11 (11C) was produced by 14N (p,α)11C nuclear reaction using the cyclotron (HM-18, Sumitomo Heavy Industry, Tokyo, Japan) at the Hamamatsu Photonics PET Center and obtained as [11C]CO2, which was converted to [11C]methyl iodide. [11C]2β-carbomethoxy-3β-(4-fluorophenyl)tropane (β-CFT) (for DAT) and [11C]SCH 23390 (for DA D1 receptors) were synthesized as previously reported (Tsukada et al, 2001; Harada et al, 2002). The radiochemical and chemical purities of labeled compounds were greater than 98 and 99%, respectively. After analysis for identification, the solution was passed through a 0.22-μm pore size filter before intravenous administration to the monkey.
PET Scans
PET data were collected before and 3 weeks after administration of MAP or MAP/NAC. Data were collected on a high-resolution PET scanner (SHR-7700, Hamamatsu Photonics KK, Hamamatsu, Japan) with a transaxial resolution of 2.6 mm full-width at half-maximum (FWHM) and a center-to-center distance of 3.6 mm (Watanabe et al, 1997). The PET camera allowed 31 slices for imaging to be recorded simultaneously. After an overnight fast, animals were fixed to the monkey chair with stereotactic coordinates aligned parallel to the OM line. A cannula was implanted in the posterior tibial vein of the monkey for administration of [11C]-labeled compounds. [11C]β-CFT or [11C]SCH 23390 was injected through the posterior tibial vein cannula. For [11C]SCH 23390, a PET scan was performed for 64 min with six time frames at 10-s intervals, six time frames at 30-s intervals, 12 time frames at 1-min intervals, followed by 16 time frames at 3-min intervals. For [11C]β-CFT, additional scans of nine time frames at 3-min intervals were carried out to collect data for 91 min total. After completion of the first scan with [11C]β-CFT, scans with [11C]SCH 23390 were continuously performed at 3-h intervals. Due to the very short half-life of 11C (20.4 min), a time lag of at least 3 h between scans provided a sufficient decay time of radioactivity in monkeys (approximately 1/400 of the injected dose). Therefore, the level of radioactivity associated with the previous injection of labeled compound would not interfere with the next scan.
Data Analysis and Statistical Analysis
For quantitative analysis, time–activity curves of radioactivity in the cerebellum, used as an input function because of the much lower density of dopamine receptors (Creese et al, 1975), and each region of interest (ROI) were fitted to a two-compartment model using the least-squares fitting method to estimate the kinetic parameters, and the binding potential in each ROI was calculated as described previously (Lammertsma and Hume, 1996). The differences between the control (pre-MAP) monkeys and the MAP or NAC plus MAP (post MAP) monkeys were determined using a paired two-tailed t-test. The difference between monkeys in the MAP-treated group and those in the NAC plus MAP-treated group was determined using an unpaired two-tailed t-test. Significance was set at p<0.05.
RESULTS
PET studies using [11C]β-CFT (for DAT) or [11C]SCH 23390 (for DA D1 receptor) were performed before and 3 weeks after repeated administration of MAP. High accumulation of radioactivity in the striatum after intravenous administration of [11C]β-CFT or [11C]SCH 23390 was detected in the control monkeys, whereas levels of radioactivity in the cerebellum were much lower compared to those in the striatum (Figure 2). At 3 weeks after administration of MAP (2 mg/kg × 4, 2-h intervals), the binding of [11C]β-CFT in the striatum was significantly (t=10.01, p=0.010) decreased, whereas the binding of [11C]SCH 23390 to DA D1 receptor in the striatum was not altered (t=0.716, p=0.549) (Figure 2 and Table 1). Time–activity curves of radioactivity after intravenous administration of [11C]β-CFT indicated a high accumulation of radioactivity in the striatum, with a low level of radioactivity in the cerebellum, of the control monkeys (Figure 3). At 3 weeks after administration of MAP (2 mg/kg × 4, 2-h intervals), time–activity curves of radioactivity in the striatum after intravenous administration of [11C]β-CFT were markedly decreased, whereas those of radioactivity in the cerebellum were not altered (Figure 3).

PET images of [11C]SCH 23390 and [11C]β-CFT in the brains of a rhesus monkey (Macaca mulatta). PET data were collected on an animal PET scanner (Hamamatsu SHR-7700) with a transaxial resolution of 2.6 mm (FWHM). The PET image of [11C]SCH23390 was generated by the summation of data from 37 to 64 min after injection. PET images for [11C]β-CFT were generated by the summation of data from 61 to 91 min after injection. The stereotactic coordinates of PET were adjusted based on the OM line. These PET images were from the same monkey.

Time–activity curves of radioactivity in the striatum and cerebellum of the monkey before and 3 weeks after administration of MAP. PET data were collected for 91 min. The radioactivity in the striatum and cerebellum was plotted against time after intravenous administration of [11C]β-CFT into a rhesus monkey.
As shown in Figure 4, the accumulation of radioactivity in the striatum of monkeys treated with NAC plus MAP after administration of [11C]β-CFT was higher than that of radioactivity in the striatum of monkeys treated with MAP alone (Figure 4). The binding potential in the striatum of monkeys treated with NAC plus MAP after administration of [11C]β-CFT was significantly (t=11.74, p<0.001) higher than that of the striatum of monkeys treated with MAP alone, although the binding potential in the striatum of monkeys treated with NAC plus MAP was significantly (t=4.367, p=0.049) lower than that of the striatum of control monkeys (Table 1).

PET images of [11C]β-CFT in the brains of a rhesus monkey 3 weeks after administration of MAP or NAC plus MAP. Each PET image was generated by the summation of data from 61 to 91 min after injection of [11C]β-CFT.
DISCUSSION
The major finding of the present study is that infusion of the antioxidant NAC could attenuate the reduction of DAT in the monkey striatum after administration of MAP. Repeated administration of MAP caused a marked reduction of DAT in the monkey striatum, which is consistent with previous reports (Melega et al, 1998, 2000; Villemagne et al, 1998). In contrast, we found that the binding of [11C]SCH 23390 to DA D1 receptors in the monkey striatum was not altered by administration of MAP, suggesting that administration of MAP could damage the dopaminergic nerve terminals but not the postsynaptic neurons. In addition, it seems that the drug schedule (2 mg/kg × 4, 2-h intervals) used in this study is neurotoxic to DAT in the monkey brain. As described in Introduction, the marked release of DA produced by MAP could be implicated in the neurotoxic effects on dopaminergic terminals in the brain after administration of MAP (Cadet et al, 2003; LaVoie and Hastings, 1999; Stokes et al, 1999). Two metabolic routes seem possible: (1) formation of 3,4-dihydroxyphenyl acetic acid (DOPAC) and hydrogen peroxide (H2O2) by monoamine oxidase (MAO), or (2) formation of reactive DA-quinones and free radicals by auto-oxidation (Asanuma et al, 2003). As DA-induced modifications of protein structure and function may result in cellular toxicity, it is likely that DA quinones produced by auto-oxidation contribute to MAP-induced neurotoxicity to dopaminergic nerve terminals, supporting the evidence of oxidative stress in this model (LaVoie and Hastings, 1999; Stokes et al, 1999). In addition, high ROS, including hydrogen peroxide, superoxide radical, and hydroxyl radical, are generated not only during the oxidation of DA but also during the decay of redox-active DA quinones, suggesting that superoxide radicals, hydrogen peroxide, and nitric oxide might be involved in the neurotoxicity of MAP (Davidson et al, 2001; Imam et al, 2001; Cadet et al, 2003).
It is well known that NAC can act as a precursor for glutathione synthesis, as well as a stimulator of the cytosolic enzymes involved in glutathione regeneration. Furthermore, NAC can act by direct reaction between its reducing thiol group and ROS. It has been shown that NAC can prevent programmed cell death in cultured neuronal cells and that NAC increases mitochondrial complex I and IV specific activities both in vitro and in vivo in synaptic mitochondrial preparations from aged mice (Banaclocha, 2001). Therefore, it should be noted that a potent antioxidant NAC could attenuate the reduction of DAT in the monkey striatum after the repeated administration of MAP.
DAT knockout mice are resistant to MAP-induced neurotoxicity of dopaminergic nerve terminals, suggesting the role of DAT in the MAP-induced neurotoxicity in these nerve terminals (Fumagalli et al, 1998). Nevertheless, it is possible that MAP-induced DA released within the cytoplasm of dopaminergic terminals might be a more critical trigger of MAP-induced neurotoxicity in the dopaminergic terminals, since vesicular monoamine transporter 2 (VMAT2) knockout mice are more susceptible to the toxic manifestations of MAP (Fumagalli et al, 1999). These findings are supported by the report that MAP-induced neurotoxicity might be related to the formation of DA quinones, a process dependent on increased DA levels within dopaminergic nerve terminals (LaVoie and Hastings, 1999). It is likely that NAC binds to reactive DA quinones and ROS by auto-oxidation, resulting in the protection of the neurotoxicity by DA quinones.
The frequency of emergency room visits due to acute MAP intoxication has increased dramatically in the past few years (Lan et al, 1998; Cadet et al, 2003). In toxic doses, MAP can cause agitation, anxiety, hallucinations, delirium, psychosis, cognitive and psychomotor impairment, seizures, and death (Lan et al, 1998; Cadet et al, 2003). NAC is currently the ‘gold-standard’ treatment approach for management of acetaminophen-induced hepatotoxicity. Furthermore, NAC appears to have some clinical usefulness as a chelating agent in the treatment of acute heavy-metal poisoning, both as an agent capable of protecting the liver and kidney from damage and as an intervention to enhance elimination of the metals (Kelly, 1998). Given these two advantages, it is likely that NAC is a useful drug for treatment of neurotoxicity in dopaminergic nerve terminals in the human brain caused by chronic use of MAP.
In conclusion, our findings demonstrate that the potent antioxidant NAC could attenuate the reduction of DAT in the monkey striatum after repeated administration of MAP. Therefore, it is possible that NAC would be a suitable drug for treatment of MAP abusers, since NAC has been widely used as a therapeutic drug or a nutritional supplement.
References
Asanuma M, Miyazaki I, Ogawa N (2003). Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson's disease. Neurotox Res 5: 165–176.
Banaclocha MM (2001). Therapeutic potential of N-acetylcysteine in age-related mitochondrial neurodegenerative diseases. Med Hypotheses 56: 472–477.
Cadet JL, Jayanthi S, Deng X (2003). Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J 17: 1775–1788.
Creese I, Burt DR, Snyder SH (1975). Dopamine receptor binding: differentiation of agonist and antagonist states with [3H]dopamine and [3H]haloperidol. Life Sci 17: 993–1002.
Davidson C, Gow AJ, Lee TH, Ellinwood EH (2001). Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Rev 36: 1–22.
Fukami G, Hashimoto K, Koike K, Okamura N, Shimizu E, Iyo M (2004). Effect of antioxidant N-acetyl-L-cysteine on behavioral changes and neurotoxicity in rats after administration of methamphetamine. Brain Res. in press.
Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG (1998). Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci 18: 4861–4869.
Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG (1999). Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci 19: 2424–2431.
Harada N, Nishiyama S, Satoh K, Fukumoto D, Kakiuchi T, Tsukada H (2002). Age-related changes in the striatal dopaminergic system in the living brain: a multiparametric PET study in conscious monkeys. Synapse 45: 38–45.
Imam SZ, El-Yazal J, Newport GD, Itzhak Y, Cadet JL, Slikker W et al (2001). Methamphetamine-induced dopaminergic neurotoxicity: role of peroxynitrite and neuroprotective role of antioxidants and peroxynitrite decomposition catalysts. Ann NY Acad Sci 939: 366–380.
Kelly GS (1998). Clinical applications of N-acetylcysteine. Altern Med Rev 3: 114–127.
Konuma K (1994). Use and abuse of amphetamines in Japan In: Cho AK, Segal DS (eds). Amphetamine and Its Analogs. Academic Press: San Diego. pp 415–435.
Lammertsma A, Hume S (1996). Simplified reference tissue model for PET receptor studies. Neuroimage 4: 153–158.
Lan KC, Lin YF, Yu FC, Lin CS, Chu P (1998). Clinical manifestations and prognostic features of acute methamphetamine intoxication. J Formos Med Assoc 97: 528–533.
LaVoie MJ, Hastings TG (1999). Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci 19: 1484–1491.
McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA (1998). Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci 18: 8417–8422.
Melega WP, Lacan G, Harvey DC, Huang SC, Phelps ME (1998). Dizocilpine and reduced body temperature do not prevent methamphetamine-induced neurotoxicity in the vervet monkey: [11C]WIN 35,428—positron emission tomography studies. Neurosci Lett 258: 17–20.
Melega WP, Lacan G, Desalles AA, Phelps ME (2000). Long-term methamphetamine-induced decreases of [11C]WIN 35,428 binding in striatum are reduced by GDNF: PET studies in the vervet monkey. Synapse 35: 243–249.
Molnar Z, Shearer E, Lowe D (1999). N-acetylcysteine treatment to prevent the progression of multisystem organ failure: a prospective, randomized, placebo-controlled study. Crit Care Med 27: 1100–1104.
Onoe H, Inoue O, Suzuki K, Tsukada H, Ito T, Magata N et al (1994). Ketamine increases the striatal N-11C-methylspiperone binding in vivo: positron emission tomography study using conscious rhesus monkey. Brain Res 663: 191–198.
Rank N, Michel C, Haertel C, Lenhart A, Welte M, Meier-Hellmann A et al (2000). N-acetylcysteine increases liver blood flow and improves liver function in septic shock patients: results of a prospective, randomized, double-blind study. Crit Care Med 28: 3799–3807.
Sekine Y, Iyo M, Ouchi Y, Matsunaga T, Tsukada H, Okada H et al (2001). Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am J Psychiatry 158: 1206–1214.
Sekine Y, Minabe Y, Ouchi Y, Takei N, Iyo M, Nakamura K et al (2003). Association of dopamine transporter loss in the orbitofrontal and dorsolateral prefrontal cortices with methamphetamine-related psychiatric symptoms. Am J Psychiatry 160: 1699–1701.
Shimizu E, Hashimoto K, Komatsu N, Iyo M (2002). Roles of endogenous glutathione levels on 6-hydroxydopamine-induced apoptotic neuronal cell death in human neuroblastoma SK-N-SH cells. Neuropharmacology 43: 434–443.
Stokes AH, Hastings TG, Vrana KE (1999). Cytotoxic and genotoxic potential of dopamine. J Neurosci Res 55: 565–659.
Takechi H, Onoe H, Imamura K, Onoe K, Kakiuchi T, Nishiyama S et al (1994). Brain activation study by use of positron emission tomography in unanesthetized monkey. Neurosci Lett 182: 279–282.
Tsukada H, Harada N, Nishiyama S, Ohba H, Sato K, Fukumoto D et al (2000). Ketamine decreased striatal [11C]raclopride binding with no alterations in static dopamine concentrations in the striatal extracellular fluid in the monkey brain: multi-parametric PET studies combined with microdialysis analysis. Synapse 37: 95–103.
Tsukada H, Miyasato K, Kakiuchi T, Nishiyama S, Harada N, Domino EF (2002). Comparative effects of methamphetamine and nicotine on the striatal [11C]raclopride binding in unanesthetized monkeys. Synapse 45: 207–212.
Tsukada H, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Harada N (2001). Ketamine alters the availability of striatal dopamine transporter as measured by [11C] β-CFT and [11C] β-CIT-FE in the monkey brain. Synapse 42: 273–280.
Tsukada H, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Harada N et al (1999). Isoflurane anesthesia enhances the inhibitory effects of cocaine and GBR12909 on dopamine transporter: PET studies in combination with microdialysis in the monkey brain. Brain Res 849: 85–96.
Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB et al (1998). Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C]WIN-35,428 positron emission tomography studies and direct in vitro determinations. J Neurosci 18: 419–427.
Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M et al (2001a). Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci 21: 9414–9418.
Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D et al (2001b). Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry 158: 377–382.
Watanabe M, Okada H, Shimizu K, Omura T, Yoshikawa E, Kosugi T et al (1997). A high resolution animal PET scanner using compact PS-PMT detectors. IEEE Trans Nucl Sci 44: 1277–1282.
Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM et al (1996). Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med 2: 699–703.
Zafarullah M, Li WQ, Sylvester J, Ahmad M (2003). Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 60: 6–20.
Acknowledgements
This work was supported in part by Grant-in-Aid for Creative Scientific Research of Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hashimoto, K., Tsukada, H., Nishiyama, S. et al. Protective Effects of N-acetyl-L-cysteine on the Reduction of Dopamine Transporters in the Striatum of Monkeys Treated with Methamphetamine. Neuropsychopharmacol 29, 2018–2023 (2004). https://doi.org/10.1038/sj.npp.1300512
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300512
Keywords
This article is cited by
-
Mechanistic Effects and Use of N-acetylcysteine in Substance Use Disorders
Current Behavioral Neuroscience Reports (2022)
-
The Potential of N-Acetyl-L-Cysteine (NAC) in the Treatment of Psychiatric Disorders
CNS Drugs (2022)
-
Epigallocatechin Gallate Mitigates the Methamphetamine-Induced Striatal Dopamine Terminal Toxicity by Preventing Oxidative Stress in the Mouse Brain
Neurotoxicity Research (2020)
-
A study protocol for the N-ICE trial: A randomised double-blind placebo-controlled study of the safety and efficacy of N-acetyl-cysteine (NAC) as a pharmacotherapy for methamphetamine (“ice”) dependence
Trials (2019)
-
Role of Mitochondria in Methamphetamine-Induced Dopaminergic Neurotoxicity: Involvement in Oxidative Stress, Neuroinflammation, and Pro-apoptosis—A Review
Neurochemical Research (2018)
