
Abstract
The enhancement of central serotonin system function underlies the therapeutic effects of selective serotonin reuptake inhibitors (SSRIs), which have become the most commonly used class of antidepressant agents. However, many individuals experience depressive episodes that are resistant to SSRI treatment. Homeostatic mechanisms that limit the extent to which SSRIs enhance serotonergic neurotransmission have been implicated in this phenomenon. Here, we report a novel strategy for enhancing the efficacy of SSRIs. Using in vivo microdialysis methods in rats, the nonselective 5-HT2 receptor antagonist ketanserin was observed to produce a robust augmentation of citalopram-, fluoxetine-, and sertraline-induced elevations of hippocampal extracellular serotonin levels. Similar effects were also observed in cortex. The potentiation of SSRI-induced increases in hippocampal serotonin levels was reproduced by the 5-HT2C receptor-selective antagonists SB 242084 and RS 102221, but not by the 5-HT2A receptor-selective antagonist MDL 100 907. Although 5-HT2C receptor antagonists augmented the actions of SSRIs, they had no effect on extracellular serotonin levels or tail suspension responses when administered alone. These results were in strong accord with independent findings using a line of 5-HT2C receptor-null mutant mice. Although this mutation did not affect baseline extracellular serotonin levels or tail suspension test (TST) behavior, it enhanced fluoxetine effects on serotonin levels and immobility in the TST. These findings reveal an unanticipated pharmacological action of 5-HT2C receptors that warrants consideration in the development of novel strategies for the treatment of depression.
Similar content being viewed by others


INTRODUCTION
Major depressive disorder is among the most common and debilitating psychiatric conditions, with a lifetime prevalence of 20% for females and 10% for males (Judd, 1995). The high incidence and chronic recurrent course of unipolar depression has led the World Health Organization to predict that it will become the second-ranking global cause of illness-related disability by 2020 (Murray and Lopez, 1997). Enhancement of central serotonin system function has been associated with the actions of a wide variety of antidepressant treatments, as indicated by the widespread use of selective serotonin reuptake inhibitors (SSRIs). The SSRIs, which are generally considered to be the first-line antidepressants of choice (Fava, 2000), block the plasma membrane serotonin reuptake transporter (SERT) and thus augment the availability of synaptic serotonin. However, this augmentation is blunted by SSRI-induced increases in serotonin levels in the vicinity of brainstem raphe nuclei serotonergic neurons. In response to elevated serotonin levels, somatodendritic 5-HT1A autoreceptors are activated, reducing serotonergic neuronal firing rates and forebrain serotonin release (Artigas et al, 1996). The characteristic delay in the onset of antidepressant effects of SSRIs has been attributed to a gradual decrease in this inhibition, resulting from partial 5-HT1A autoreceptor desensitization (Hervas et al, 2001).
These findings led to the proposal that the coadministration of SSRIs with 5-HT1A receptor antagonists could result in accelerated and possibly enhanced antidepressant responses (Artigas, 1993). This hypothesis was examined in clinical studies using the nonselective 5-HT1A receptor antagonist pindolol. There was some indication that the onset of antidepressant responses was accelerated by the combined pharmacotherapy, although results were mixed (Artigas et al, 2001; Brousse et al, 2003). Since pindolol has partial 5-HT1A receptor agonist activity, along with very high affinity at β-adrenoceptors, the receptor mechanisms through which pindolol influences antidepressant action have been called into question (Artigas et al, 2001; Cremers et al, 2001; Brousse et al, 2003). Thus, clinical tests of this antidepressant augmentation strategy await the availability of potent and selective 5-HT1A receptor antagonist compounds.
The 5-HT2C receptor is another 5-HT receptor subtype that warrants consideration in the development of novel treatment strategies for depression. Although a wide variety of antidepressants have high-affinity antagonist activity at 5-HT2C receptors (Jenck et al, 1994; Palvimaki et al, 1996), the contributions of this activity to their therapeutic effects are poorly understood. Indirect activation of 5-HT2C receptors by SSRI treatment may contribute to the transient anorectic and anxiogenic effects of these compounds (Halford and Blundell, 2000; Bagdy et al, 2001). However, the contribution of these receptors to the neurochemical and antidepressant behavioral effects of SSRIs remains unclear. Recent findings indicating that RNA editing of these receptors is regulated by SSRI treatment and altered in the prefrontal cortex of suicide victims highlights the importance of clarifying the contributions of these receptors to antidepressant action (Niswender et al, 2001; Gurevich et al, 2002a; Gurevich et al, 2002b).
Here, we report a novel interaction through which pharmacological blockade of 5-HT2C receptors potentiates both the neurochemical and the antidepressant-like behavioral actions of SSRIs. We also report independently obtained data revealing that the effects of a 5-HT2C receptor-null mutation on neurochemical and behavioral responses to SSRI administration are highly concordant with those of 5-HT2C receptor antagonist treatment. These results suggest a potential treatment strategy that may counter SSRI-induced negative feedback of serotonergic neurotransmission and augment the antidepressant actions of these drugs.
MATERIALS AND METHODS
Animals
All animals were group-housed at 22°C on a 12-h light/dark cycle, with standard food and water ad libitum. Adult male albino rats of a Wistar-derived strain (285–320 g; Harlan, Zeist, Netherlands) were used. Effects of 5-HT2C receptor antagonism in the mouse tail suspension test were determined using C57BL/6 mice (M & B A/S, Denmark). To examine the effects of genetic inactivation of 5-HT2C receptors, male 8–12-week old mice hemizygous for a null mutation of the X-linked htr2c gene (backcrossed to a C57BL/6J background for at least 24 generations) and wild-type littermates were used (Tecott et al, 1995). The line has been maintained through matings of heterozygous females with C57BL/6J males (Jackson Laboratory, Bar Harbor, ME). Experiments were conducted in accordance with the Declaration of Helsinki and the NIH Guide for the Care and Use of Laboratory Animals.
Drugs
For rat studies, citalopram hydrobromide, fluoxetine hydrochloride, irindalone, sertraline hydrochloride, olanzapine, RS 102221, and SB 242084 were obtained from Lundbeck A/S (Copenhagen, Denmark). Mepyramine, prazosin, and ketanserin were obtained from RBI (Natick, USA). MDL 100907 was synthesized at the University of Groningen. These drugs were dissolved in saline and injected subcutaneously in a volume of 1 ml/kg. Antagonists were administered immediately (within 15 s) prior to either SSRI or vehicle treatment. For mutant mouse studies, fluoxetine hydrochloride was obtained from Tocris (Ellisville, MO). Fluoxetine was dissolved in a sterile saline solution for i.p. injections. Control injections consisted of vehicle only.
Surgical Implantation of Microdialysis Probes
Rat microdialysis studies were performed at the University of Groningen, The Netherlands. For rats, microdialysis was performed using I-shaped probes, composed of polyacrylonitrile/sodium methyl sulfonate copolymer dialysis fiber (i.d. 220 μm, o.d. 310 μm, AN 69, Hospal, Italy). The exposed length of the membranes was 4 mm for both ventral hippocampus and prefrontal cortex. Prior to surgery, rats were anaesthetized using ketamine (50 mg/kg i.p.) and xylazine (8 mg/kg i.p.), after premedication with midazolam (5 mg/kg s.c.). Lidocaine-HCl (10% (m/v)) was used for local anaesthesia. Rats were placed in a stereotaxic frame (Kopf, USA), and probes were implanted into the ventral hippocampus (L +4.8 mm, IA: +3.7 mm, V: −8.0 mm) and prefrontal cortex (L –0.9 mm, AP: +3.5 mm, V: −6.0 mm). After insertion, probes were secured with dental cement.
Microdialysis studies utilizing 5-HT2C receptor mutant mice were performed at the University of California, San Francisco. For mice, concentric microdialysis probes were constructed using 23 gauge stainless steel and silica capillary tubing. The dialysis membrane (i.d. 240 μm, o.d. 290 μm, AN 69HF, Hospal, Italy) consisted of polyacrilonitril/sodium methyl sulfonate copolymer with an average pore size of 29 A. Dialysis probes had 2 mm of exposed membrane. Prior to surgery, mice were anesthetized using ketamine (100 mg/kg i.p.) and xylazine (10 mg/kg i.p.) and then placed in a stereotaxic frame. Microdialysis probes were implanted into the right prefrontal cortex (PFC) at the following coordinates relative to bregma (AP +2 mm; L −0.5 mm; DV –4 mm; (Franklin and Paxinos, 1997)).
In Vivo Microdialysis
All animals used in microdialysis studies were allowed to recover for at least 24 h following surgery. For rat studies, probes were perfused with artificial cerebrospinal fluid containing (in mM): NaCl 147, KCl 3.0, CaCl2 1.2, and MgCl2 1.2, at a flow-rate of 1.5 μl/min (Harvard Apparatus pump, South Natick, MA, USA). Microdialysis samples were collected at 15 min intervals in HPLC vials containing 7.5 μl 0.02 M acetic acid for serotonin analysis (animals were injected at t=0, data were not corrected for lag time). For mouse studies, probes were perfused with artificial cerebrospinal fluid containing (in mM): NaCl 145, KCl 2.7, MgCl2 1, CaCl2 1.2, Na2HPO4 2, pH 7.40, at a flow rate of 1 μl/min. Microdialysis samples were collected at 20 min intervals. Fluoxetine was administered immediately before sample 6.
Determination of Dialysate Serotonin Levels
Serotonin concentrations were determined using HPLC coupled with electrochemical detection. For rat studies, 20 μl microdialysate fractions were injected via an autoinjector (CMA/200 refrigerated microsampler, CMA, Sweden) onto a 100 × 2.0 mm C18 Hypersil 3 μm column (Bester, Amstelveen, the Netherlands) and separated with a mobile phase consisting of 5 g/l di-ammoniumsulfate, 500 mg/l Na2-EDTA, 50 mg/l heptane sulfonic acid, 4% methanol v/v, and 30 μl/l of triethylamine, pH 4.75 at a flow rate of 0.4 ml/min (Shimadzu LC-10 AD). 5-HT was detected amperometrically at a glassy carbon electrode at 500 mV vs Ag/AgCl (Antec Leyden, Leiden, Netherlands). The detection limit was 0.5 fmol 5-HT per 20 μl sample (signal-to-noise ratio 3).
For mouse studies, the mobile phase consisted of a mixture of acetonitrile, phosphate buffer, and an ion-pairing agent (ESA, Chelmsford, MA). The mobile phase was delivered by a pump (model 582, Solvent Delivery Module; ESA) at a flow rate of 0.25 μl/min through an MD-150 narrowbore column (ESA). 5-HT was detected using a coulometric detector (Coulochem II; ESA) coupled to a single channel enhanced amperometric cell (Model 5041; ESA). The potential applied to the electrode was +175 mV; under these conditions, the limit of detection for 5-HT was approximately 2–3 fmol. Data were analyzed using a basic integrator (Model 3395; Hewlett-Packard).
For microdialysis data, basal 5-HT levels were defined as the average level of the first five samples. Differences between treatments were analyzed using a two-way ANOVA with repeated measurements, followed by Students–Newman–Keuls post hoc analysis. In separate analyses for evaluating the effects of treatments relative to baseline, one-way ANOVA for repeated measurements on ranks were used, followed by Dunnet's test. Level of significance was set at p⩽0.05.
Tail Suspension Test (TST)
The mouse tail suspension test was designed as a screening assay for antidepressant compounds, and is sensitive to the major classes of antidepressants (Steru et al, 1985; Porsolt, 2000). TST studies employing the 5-HT2C receptor antagonist SB 206553 were performed at Lundbeck A/S in Copenhagen, Denmark. TST studies utilizing 5-HT2C receptor mutant mice were performed at the University of California, San Francisco. Animals were suspended by the tail using adhesive tape from either a 1.2-cm diameter metal bar (5-HT2C receptor mutant studies) or a 1 cm diameter hook (5-HT2C receptor antagonist studies) elevated 30–35 cm. When suspended, rodents either make apparent escape attempt movements or adopt a characteristic immobile posture. The total immobility time was measured (manually for 5-HT2C receptor mutant studies, and by automated strain gauge for 5-HT2C receptor antagonist studies (Med Associates, St Albans, VT)) during the 6 min test period. The effects of treatments relative to baseline were analyzed using a one-way ANOVA, followed by Dunnet's test. Interactions between antagonist treatment and SSRI treatment, or between genotype and SSRI treatment, were analyzed by a two-way ANOVA.
RESULTS
Potentiation of SSRI-Induced Serotonin Release by Ketanserin
As part of an effort to probe the contribution of serotonin receptor subtypes to the regulation of brain serotonin systems and neurochemical responses to SSRIs, the effect of coadministration of the nonselective 5-HT2 receptor antagonist ketanserin on SSRI-induced serotonin release was examined in rats using in vivo microdialysis. Microdialysis probes were placed in the ventral hippocampal formation, where baseline extracellular serotonin levels were 4.81±0.38 fmol/sample (n=95). Whereas the SSRI citalopram (3 mg/kg s.c.) produced 400–500% increases in serotonin levels relative to baseline (X210=35.5, p<0.0001), ketanserin administration (40 μg/kg s.c.) alone produced no effect (Figure 1a). However, the effect of citalopram on extracellular serotonin was substantially augmented, in a dose-dependent manner, by coadministration with ketanserin. Whereas 0.4 μg/kg ketanserin was devoid of any effect, 4 and 40 μg/kg augmented the effect of citalopram, with the 40 μg/kg dose producing 1000% increases from baseline values (F(1,106)=15.82 p<0.005; Supplemental Figure 1). A parallel set of studies using another partially selective 5-HT2 receptor antagonist, irindalone, yielded similar results (data not shown).

Augmentation of SSRI-induced serotonin release in hippocampus (a–c) and prefrontal cortex (d) of freely moving rats by coadministration of the 5-HT2 receptor antagonist ketanserin. All drugs were administered at time 0. (a) Citalopram administration (3 mg/kg s.c.) significantly increased extracellular serotonin levels in the ventral hippocampus (X210=35.5, p<0.0001, n=7). Ketanserin (40 μg/kg s.c.) did not alter hippocampal serotonin levels when administered alone (n=4). However, when coadministered with citalopram, it augmented the serotonin-releasing effects of the SSRI (F(1,106)=15.82, **p<0.005, n=4). (b) Fluoxetine administration (4 mg/kg s.c.) significantly increased hippocampal extracellular serotonin levels (X210=34.3, p<0.0005, n=5). Ketanserin (40 μg/kg s.c.) augmented the serotonin-releasing effects of fluoxetine (F(1,108)=6.44, *p<0.05, n=5). (c) Sertraline administration (9 mg/kg s.c.) significantly increased hippocampal serotonin extracellular levels (X210=33.0, p<0.0005, n=4). Ketanserin (40 μg/kg s.c.) augmented the serotonin-releasing effects of sertraline (F(1,87)=4.19, ***p<0.0005, n=4). (d) In the PFC, citalopram administration (3 mg/kg s.c.) significantly increased extracellular serotonin levels (X210=68.7, p<0.0001, n=10). As observed in the hippocampus, ketanserin (40 μg/kg s.c.) alone produced no effect (n=7), but it significantly augmented the effect of citalopram (3 mg/kg s.c.) (F(1,163)=6.27, *p<0.05, n=4).
We then determined the extent to which the ability of ketanserin to potentiate citalopram-induced serotonin release generalizes to additional SSRIs. Whereas fluoxetine alone (4 mg/kg) increased serotonin levels to approximately 350% of the baseline value (X210=34.3, p<0.0005), ketanserin coadministration (40 μg/kg s.c.) augmented this effect to produce a 600% increase relative to baseline values (F(1,108)=6.44, p<0.05) (Figure 1b). Sertraline (9 mg/kg, s.c.) also increased serotonin levels (X210=33.0, p<0.0005) and ketanserin (40 μg/kg, s.c.) potentiated this effect (F(1,87)=4.19, p<0.0005) (Figure 1c). To determine whether the ability of ketanserin to potentiate citalopram-induced serotonin release generalizes to other forebrain regions implicated in the regulation of affect, the experiment was repeated with microdialysis probes located in the prefrontal cortex (PFC). In this region, baseline extracellular serotonin levels were 3.79±0.37 fmol/sample (mean±SEM, n=19). Citalopram administration (3 mg/kg s.c.) increased extracellular serotonin levels in the PFC (X210=68.7, p<0.0001) and ketanserin potentiated this effect (F(1,163)=6.27, p<0.05) (Figure 1d).
Identification of Receptor Subtype Mediating Potentiation of SSRI-Induced Serotonin Release
Ketanserin interacts with high affinity at 5-HT2 receptors and at several non-5-HT receptor subtypes (Roth et al, 2000). To determine the extent to which these receptors contribute to the ability of ketanserin to potentiate SSRI-induced serotonin release, antagonists of these receptors were coadministered with citalopram.
The α1-adrenoceptor antagonist prazosin (0.4 mg/kg s.c.) significantly decreased 5-HT levels when administered alone (X210=19.9, p<0.05), but did not alter the effect of citalopram (Figure 2a). The histamine H1-receptor antagonist mepyramine (0.3 mg/kg s.c.) did not alter 5-HT levels when administered alone, nor did it modify the effect of citalopram (Figure 2b). Similarly, the selective 5-HT2A receptor antagonist MDL 100,907 (0.4 mg/kg s.c.) had no effect on 5-HT levels when administered alone or in combination with citalopram (Figure 2c). In contrast, two selective 5-HT2C receptor antagonists, SB 242084 and RS 102221, potentiated citalopram-induced serotonin release. Although these agents did not produce significant alterations of extracellular serotonin levels when given alone (0.4 and 0.6 mg/kg s.c., respectively), they substantially augmented the serotonergic response to citalopram, as indicated by 900% increases of extracellular serotonin levels from baseline values (SB 242084: F(1,117)=9.19, p<0.05; RS 102221: F(1,120)=12.14, p<0.005; Figures 3a, b).

SSRI-induced hippocampal serotonin release in freely moving rats is not potentiated by coadministration of an α1-adrenoceptor antagonist, a histamine receptor 1 antagonist, or a 5-HT2A receptor antagonist. All drugs were administered at time 0. (a) Administration of the α1-adrenoceptor antagonist prazosin (0.4 mg/kg s.c.) alone significantly reduced 5-HT extracellular levels (X210=19.9, p<0.05, n=4). However, prazosin did not alter serotonin release induced by citalopram (3 mg/kg s.c.; coadministration n=8, citalopram alone n=4). (b) Administration of the H1 receptor antagonist mepyramine (0.3 mg/kg s.c.) did not alter extracellular serotonin levels when administered alone (n=5) or in combination with citalopram (3 mg/kg s.c.; coadministration n=7, citalopram alone n=7). (c) Administration of the selective 5-HT2A receptor antagonist MDL 100,907 (0.4 mg/kg s.c.) did not alter extracellular serotonin levels when administered alone (n=5) or in combination with citalopram (3 mg/kg s.c.; coadministration n=5, citalopram alone n=7).

Coadministration of 5-HT2C receptor antagonists potentiated SSRI-induced hippocampal serotonin release in freely moving rats. All drugs were administered at time 0. (a) Whereas the selective 5-HT2C receptor antagonist SB 242084 (0.4 mg/kg s.c.) did not alter serotonin levels when administered alone (n=5), it significantly augmented the serotonergic neurochemical response to 3 mg/kg s.c. citalopram (F(1,117)=9.19, *p<0.05, n=5; citalopram alone n=7). (b) Another 5-HT2C receptor antagonist, RS 102221 (0.6 mg/kg s.c.) did not alter serotonin levels when administered alone (n=5), but significantly augmented the serotonergic neurochemical response to 3 mg/kg s.c. citalopram (F(1,120)=12.14, **p< 0.005, n=5; citalopram alone n=7).
Effect of htr2c Gene Inactivation on Neurochemical Responses to Fluoxetine
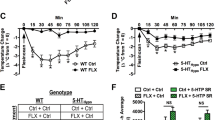
To examine the contributions of 5-HT2C receptors to the actions of SSRIs, we also used a mouse molecular genetic approach. An in vivo microdialysis study was performed to examine the effect of fluoxetine (20 mg/kg, i.p.) on extracellular serotonin levels in the PFC of 5-HT2C receptor-null mutant mice (Figure 4). No phenotypic effects on basal extracellular serotonin levels were observed (5.8±0.7 and 5.6±0.6 fmol/sample (mean±SEM) in the PFC of mutant and wild-type mice, respectively). By contrast, the mutation significantly augmented the effect of fluoxetine on extracellular serotonin levels in the PFC (F(1,18)=6.018, p<0.05).

Neurochemical effects of fluoxetine are potentiated in 5-HT2C receptor null mutant mice. Fluoxetine was administered at time 0. Basal extracellular serotonin levels were (mean±SEM) 5.8±0.7 fmol/sample in the PFC of 5-HT2C receptor mutant mice and 5.6±0.6 fmol/sample in the PFC of wild-type animals and did not significantly differ between the two groups. Fluoxetine-induced (18 mg/kg i.p.) increases in serotonin levels were potentiated in mutant mice (F(1,18)=6.018, *p<0.05, n=9 WT and 11 MT).
Effect of htr2c Gene Inactivation and 5-HT2C Receptor Antagonist treatment on Tail Suspension Responses
If inactivation of 5-HT2C receptors potentiates antidepressant-like behavioral responses to SSRIs, then we would expect the effects of fluoxetine to be augmented in 5-HT2C receptor mutant mice during tail suspension testing. Saline-treated mutant and wild-type mice displayed similar immobility times. Fluoxetine treatment (20 mg/kg i.p., 40 min prior to testing) significantly enhanced immobility, revealed by two-way ANOVA as a significant overall effect of drug treatment (F(1,27)=139, p<0.001). This effect of fluoxetine was potentiated in 5-HT2C receptor mutant mice, as indicated by a significant genotype × drug interaction (F(3,27)=8.7, p<0.01; Figure 5a). In an independently conducted experiment, a similar effect was observed in wild-type mice treated with the 5-HT2C receptor antagonist SB 206553. Although neither citalopram nor SB 206553 produced significant effects on immobility, coadministration of these compounds (SB 206553 5 mg/kg, s.c., 30 min prior to testing; citalopram 16 mg/kg s.c.) produced a significant anti-immobility response (F(3,28)=4.35, p<0.02) (Figure 5b).

Potentiation of behavioral effects of citalopram and fluoxetine by 5- HT2C receptor inactivation. (a) Immobility times of 5-HT2C receptor mutant and wild-type mice during tail suspension assay. No significant effect of genotype on immobility time was observed in saline-treated animals. However, fluoxetine administration (20 mg/kg i.p.) elicited a significantly greater effect on immobility time in 5-HT2C receptor mutant mice than in wild-type controls (F(1,27)=8.7, **p<0.01; n=6–10 mice per group). (b) Coadministration of a 5-HT2C receptor antagonist potentiates the effect of fluoxetine on immobility time in the tail suspension test. Administration of the 5-HT2C receptor antagonist SB 206553 (5 mg/kg s.c.) produced no significant alteration in behavioral responses to tail suspension. However, when coadministered with citalopram (16 mg/kg s.c.), SB 206553 significantly potentiated the anti-immobility effects of the SSRI (F(3,28)=4.35, *p<0.01, n=8 per group).
DISCUSSION
Our findings indicate that serotonin 5-HT2C receptors have substantial influence on both neurochemical and antidepressant-like behavioral responses to SSRIs. The observed inability of ketanserin administered alone to alter central serotonin levels was consistent with previous studies indicating that 5-HT2 receptor agonists and antagonists produce at most small effects on serotonin levels when administered alone (Gobert and Millan, 1999; Gobert et al, 2000). We were therefore surprised to find that ketanserin produced a robust augmentation of citalopram-induced elevations of serotonin levels. This effect of ketanserin was also observed with the SSRIs fluoxetine and sertraline, indicating a general ability of ketanserin to potentiate the neurochemical effects of serotonin reuptake blockade.
Determination of the mechanism through which ketanserin potentiates SSRI action required consideration of the potential contributions of the multiple receptor subtypes for which the ketanserin has substantial antagonist affinity, such as the 5-HT2A, 5-HT2C, histamine H1, and α1-adrenergic receptors (Leysen et al, 1992; Roth et al, 2000). When corresponding subtype-selective antagonists were coadministered with citalopram, only the 5-HT2C receptor antagonists SB 242084 and RS 102221 reproduced the effects of ketanserin. However, neither antagonist altered extracellular serotonin levels when administered alone.
The potentiation of SSRI-induced serotonin release by 5-HT2C receptor inactivation was observed in both the hippocampal formation and prefrontal cortex, two regions that have been strongly implicated in the pathophysiology of depressive disorders and in responses to antidepressant drugs (Stockmeier, 1997; Davidson et al, 2002). The potential antidepressant efficacy of drug treatments has been associated with reductions of immobility time in a tail suspension assay (Steru et al, 1985; Porsolt, 2000). Since the enhancement of brain serotonin function is thought to be central to the antidepressant effects of SSRIs, we hypothesized that the antidepressant-like behavioral effects of SSRIs would be potentiated by 5-HT2C receptor inactivation. Accordingly, pharmacological blockade of 5-HT2C receptors produced a small potentiation of citalopram's effect in the TST, yet produced no observable effects in this test in the absence of SSRIs. Additional studies are required to determine the extent to which behavioral responses to the pharmacological blockade of 5-HT2C receptors generalizes to additional SSRIs.
Similar phenomena were uncovered independently by one of our groups (Tecott, American), in studies employing a line of mice bearing a null mutation of the 5-HT2C receptor gene. As this work was performed independently, a number of procedural differences exist between the European and American data sets. These include the use of different species and SSRIs for examining influences of 5-HT2C receptors. Thus, 5-HT2C receptor influences on extracellular serotonin levels were examined using 5-HT2C receptor antagonists in citalopram-treated rats (European group) and using 5-HT2C receptor mutant mice treated with fluoxetine (American group). Analogously, mouse tail suspension testing was performed using 5-HT2C receptor antagonists in citalopram-treated animals (European group) and using 5-HT2C receptor mutant mice treated with fluoxetine (American group). However, despite these procedural differences, a striking degree of concordance exists between the results obtained independently by our groups: (1) both the 5-HT2C receptor mutation and the 5-HT2C receptor antagonists enhance fluoxetine-induced extracellular serotonin levels in prefrontal cortex, (2) neither the mutation nor the antagonists themselves alter extracellular serotonin levels. Taken together, these complementary pharmacological and genetic studies provide strong evidence for the potentiating effects of 5-HT2C receptor inactivation on SSRI action.
Prior investigations of the influence of 5-HT2C receptors in assays of ‘behavioral despair’ have yielded inconsistent results. In one previous study, several 5-HT2 receptor agonist and antagonist compounds failed to influence immobility in the mouse forced swim test (Redrobe and Bourin, 1997). By contrast, in a study utilizing the rat forced swim test, several 5-HT2C receptor agonists, but not mCPP, reduced immobility in the forced swim test; these effects were blocked by 5-HT2C receptor antagonists (Cryan and Lucki, 2000). Differences between these findings and those in the present study may relate to differences in the assays (forced swim vs TST), drug doses, and the species (rats vs mice) used.
The observed influence of 5-HT2C receptors on extracellular serotonin levels during SSRI treatment, but not under baseline conditions was not anticipated. The possibility that 5-HT2 receptor antagonism could influence forebrain serotonin levels had been suggested by a prior study with the nonspecific 5-HT2C receptor antagonist mianserin (Kreiss and Lucki, 1995). However, the high affinity of mianserin for 5-HT2C receptors, α adrenoceptors, and histamine receptors precludes attribution of this effect to its activity at 5-HT2C receptors (NIMH Psychoactive Drug Screening Program Database: http://kidb.cwru.edu/pdsp.php). To our knowledge, 5-HT2C receptors have not been demonstrated on the presynaptic elements of serotonergic neurons and have not been considered to have a classic autoreceptor function. The ability of both pharmacological and genetic inactivation of 5-HT2C receptors to alter serotonin levels during SSRI treatment, but not under baseline conditions, suggests that 5-HT2C receptors may contribute to a negative feedback mechanism recruited under conditions of elevated serotonergic tone. Such a role for these receptors in the homeostatic regulation of serotonin levels could account for prior observations that 5-HT2C receptor inactivation enhances sensitivity to the motoric effects of the serotonin-releasing agents dexfenfluramine and MDMA (Vickers et al, 1999; Bankson and Cunningham, 2002).
Several possible mechanisms could underlie the augmentation of SSRI-induced serotonin release by 5-HT2C receptor inactivation. Although an autoreceptor function has not been demonstrated for 5-HT2C receptors, it is notable that these receptors are expressed in the vicinity of mesencephalic raphe serotonergic neurons that project widely to the forebrain (Clemett et al, 2000). Neurochemical and electrophysiological studies indicate that these neurons are subject to GABAergic inhibitory regulation (Gervasoni et al, 2000; Tao and Auerbach, 2003). Notably, nonspecific 5-HT receptor agonists suppress serotonin neuronal firing rates (Boothman et al, 2003), and bath application of serotonin to mesencephalic slices increases the frequency of inhibitory postsynaptic potentials in these serotonergic neurons (Liu et al, 2000). These effects are attenuated by 5-HT2A receptor antagonists, and to a much lesser extent, by 5-HT2C receptor antagonists (Liu et al, 2000; Martin-Ruiz et al, 2001; Boothman et al, 2003). It is therefore possible that functional blockade of 5-HT2C receptors in the DRN could reduce the magnitude of serotonin-mediated excitation of raphe GABA neurons. Alternatively, it is possible that serotonin release could be influenced by 5-HT2C receptors expressed within interneurons of the PFC and hippocampus (Wright et al, 1995). Nonspecific 5-HT2 receptor agonists have been found to enhance GABAergic neurotransmission in both the prefrontal cortex and CA1 region of the hippocampus (Shen and Andrade, 1998; Abi-Saab et al, 1999). The relative contributions of the 5-HT2A and 5-HT2C receptor subtypes to these effects remain to be determined.
Altogether, our results define a novel action of 5-HT2C receptors in the modulation of serotonin levels within brain regions implicated in the regulation of affect. These results further suggest that coadministration of 5-HT2C receptor antagonists with SSRIs warrants consideration as a novel approach for overcoming homeostatic mechanisms that attenuate the therapeutic efficacy of SSRIs. In this regard, additional studies are now underway to determine whether this potentiation of SSRI action is maintained during chronic coadministration with 5-HT2C receptor antagonists.
References
Abi-Saab WM, Bubser M, Roth RH, Deutch AY (1999). 5-HT2 receptor regulation of extracellular GABA levels in the prefrontal cortex. Neuropsychopharmacology 20: 92–96.
Artigas F (1993). 5-HT and antidepressants: new views from microdialysis studies. Trends Pharmacol Sci 14: 262.
Artigas F, Celada P, Laruelle M, Adell A (2001). How does pindolol improve antidepressant action? Trends Pharmacol Sci 22: 224–228.
Artigas F, Romero L, de Montigny C, Blier P (1996). Acceleration of the effect of selected antidepressant drugs in major depression by 5-HT1A antagonists. Trends Neurosci 378–383.
Bagdy G, Graf M, Anheuer ZE, Modos EA, Kantor S (2001). Anxiety-like effects induced by acute fluoxetine, sertraline or m-CPP treatment are reversed by pretreatment with the 5-HT2C receptor antagonist SB-242084 but not the 5-HT1A receptor antagonist WAY-100635. Int J Neuropsychopharmacol 4: 399–408.
Bankson MG, Cunningham KA (2002). Pharmacological studies of the acute effects of (+)-3,4-methylenedioxymethamphetamine on locomotor activity: role of 5-HT(1B/1D) and 5-HT(2) receptors. Neuropsychopharmacology 26: 40–52.
Boothman LJ, Allers KA, Rasmussen K, Sharp T (2003). Evidence that central 5-HT(2A) and 5-HT(2B/C) receptors regulate 5-HT cell firing in the dorsal raphe nucleus of the anaesthetised rat. Br J Pharmacol 139: 998–1004.
Brousse G, Schmitt A, Chereau I, Eschalier A, Dubray C, Llorca PM (2003). Interest of the use of pindolol in the treatment of depression: review. Encephale 29: 338–350.
Clemett DA, Punhani T, Duxon MS, Blackburn TP, Fone KC (2000). Immunohistochemical localisation of the 5-HT2C receptor protein in the rat CNS. Neuropharmacology 39: 123–132.
Cremers TI, Wiersma LJ, Bosker FJ, den Boer JA, Westerink BH, Wikstrom HV (2001). Is the beneficial antidepressant effect of coadministration of pindolol really due to somatodendritic autoreceptor antagonism? Biol Psychiatry 50: 13–21.
Cryan JF, Lucki I (2000). Antidepressant-like behavioral effects mediated by 5-hydroxytryptamine(2C) receptors. J Pharmacol Exp Ther 295: 1120–1126.
Davidson RJ, Pizzagalli D, Nitschke JB, Putnam K (2002). Depression: perspectives from affective neuroscience. Annu Rev Psychol 53: 545–574.
Fava M (2000). New approaches to the treatment of refractory depression. J Clin Psychiatry 61(Suppl 1): 26–32.
Franklin KJ, Paxinos G (1997). The Mouse Brain in Stereotaxic Coordinates. Academic Press Inc.: San Diego, CA.
Gervasoni D, Peyron C, Rampon C, Barbagli B, Chouvet G, Urbain N et al (2000). Role and origin of the GABAergic innervation of dorsal raphe serotonergic neurons. J Neurosci 20: 4217–4225.
Gobert A, Millan MJ (1999). Serotonin (5-HT)2A receptor activation enhances dialysate levels of dopamine and noradrenaline, but not 5-HT, in the frontal cortex of freely-moving rats. Neuropharmacology 38: 315–317.
Gobert A, Rivet JM, Lejeune F, Newman-Tancredi A, Adhumeau-Auclair A, Nicolas JP et al (2000). Serotonin(2C) receptors tonically suppress the activity of mesocortical dopaminergic and adrenergic, but not serotonergic, pathways: a combined dialysis and electrophysiological analysis in the rat. Synapse 36: 205–221.
Gurevich I, Englander MT, Adlersberg M, Siegal NB, Schmauss C (2002a). Modulation of serotonin 2C receptor editing by sustained changes in serotonergic neurotransmission. J Neurosci 22: 10529–10532.
Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C (2002b). Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron 34: 349–356.
Halford JC, Blundell JE (2000). Separate systems for serotonin and leptin in appetite control. Ann Med 32: 222–232.
Hervas I, Vilaro MT, Romero L, Scorza MC, Mengod G, Artigas F (2001). Desensitization of 5-HT(1A) autoreceptors by a low chronic fluoxetine dose effect of the concurrent administration of WAY-100635. Neuropsychopharmacology 24: 11–20.
Jenck F, Moreau JL, Mutel V, Martin JR (1994). Brain 5-HT1C receptors and antidepressants. Prog Neuro-Psychopharmacol Biol Psych 18: 563–574.
Judd LL (1995). Mood disorders in the general population represent an important and worldwide public health problem. Int Clin Psychopharmacol 10(Suppl 4): 5–10.
Kreiss DS, Lucki I (1995). Effects of acute and repeated administration of antidepressant drugs on extracellular levels of 5-hydroxytryptamine measured in vivo. J Pharmacol Exp Ther 274: 866–876.
Leysen JE, Janssen PM, Gommeren W, Wynants J, Pauwels PJ, Janssen PA (1992). In vitro and in vivo receptor binding and effects on monoamine turnover in rat brain regions of the novel antipsychotics risperidone and ocaperidone. Mol Pharmacol 41: 494–508.
Liu R, Jolas T, Aghajanian G (2000). Serotonin 5-HT(2) receptors activate local GABA inhibitory inputs to serotonergic neurons of the dorsal raphe nucleus. Brain Res 873: 34–45.
Martin-Ruiz R, Puig MV, Celada P, Shapiro DA, Roth BL, Mengod G et al (2001). Control of serotonergic function in medial prefrontal cortex by serotonin-2A receptors through a glutamate-dependent mechanism. J Neurosci 21: 9856–9866.
Murray CJ, Lopez AD (1997). Alternative projections of mortality and disability by cause 1990–2020: global burden of disease study. Lancet 349: 1498–1504.
Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, Stockmeier CA et al (2001). RNA editing of the human serotonin 5-HT2C receptor. alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology 24: 478–491.
Palvimaki EP, Roth BL, Majasuo H, Laakso A, Kuoppamaki M, Syvalahti E et al (1996). Interactions of selective serotonin reuptake inhibitors with the serotonin 5-HT2C receptor. Psychopharmacology 126: 234–240.
Porsolt RD (2000). Animal models of depression: utility for transgenic research. Rev Neurosci 11: 53–58.
Redrobe JP, Bourin M (1997). Partial role of 5-HT2 and 5-HT3 receptors in the activity of antidepressants in the mouse forced swimming test. Eur J Pharmacol 325: 129–135.
Roth BL, Kroeze WK, Patel S, Lopez E (2000). The multiplicity of serotonin receptors: uselessly diverse molecules or an embarrasment of riches? Neuroscientist 6: 252–262.
Shen RY, Andrade R (1998). 5-Hydroxytryptamine2 receptor facilitates GABAergic neurotransmission in rat hippocampus. J Pharmacol Exp Ther 285: 805–812.
Steru L, Chermat R, Thierry B, Simon P (1985). The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85: 367–370.
Stockmeier CA (1997). Neurobiology of serotonin in depression and suicide. Ann N Y Acad Sci 836: 220–232.
Tao R, Auerbach SB (2003). Influence of inhibitory and excitatory inputs on serotonin efflux differs in the dorsal and median raphe nuclei. Brain Res 961: 109–120.
Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF et al (1995). Eating disorder and epilepsy in mice lacking 5HT2C serotonin receptors. Nature 374: 542–546.
Vickers SP, Clifton PG, Dourish CT, Tecott LH (1999). Reduced satiating effect of d-fenfluramine in serotonin 5-HT2C receptor mutant mice. Psychopharmacology 143: 309–314.
Wright DE, Seroogy KB, Lundgren KH, Davis BM, Jennes L (1995). Comparative localization of serotonin1A,1C, and 2 receptor subtype mRNAs in rat brain. J Comp Neurol 351: 357–373.
Acknowledgements
We thank Odette P Murphy for technical assistance and Dr Kevin Delucchi for advice regarding statistical analysis. LT is supported by a Staglin Music Festival Investigator Award from the National Alliance for Research on Schizophrenia and Affective Disorders.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Cremers, T., Giorgetti, M., Bosker, F. et al. Inactivation of 5-HT2C Receptors Potentiates Consequences of Serotonin Reuptake Blockade. Neuropsychopharmacol 29, 1782–1789 (2004). https://doi.org/10.1038/sj.npp.1300474
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300474
Keywords
This article is cited by
-
New arylpiperazine derivatives with antidepressant-like activity containing isonicotinic and picolinic nuclei: evidence for serotonergic system involvement
Naunyn-Schmiedeberg's Archives of Pharmacology (2019)
-
A short history of the 5-HT2C receptor: from the choroid plexus to depression, obesity and addiction treatment
Psychopharmacology (2017)
-
Long-term consequences of chronic fluoxetine exposure on the expression of myelination-related genes in the rat hippocampus
Translational Psychiatry (2015)
-
Treatment-resistant depression: are animal models of depression fit for purpose?
Psychopharmacology (2015)
-
5-HT2CR Blockade in the Amygdala Conveys Analgesic Efficacy to SSRIs in a Rat Model of Arthritis Pain
Molecular Pain (2013)
