
Abstract
The Friedreich’s ataxia locus (FRDA) maps on chromosome 9q13. Genetic data, obtained from a small number of recombination events, indicated that the FRDA locus might be located centromeric to the D9S15/D9S5 linkage group, the most probable order being cen-FRDA-D9S5-D9S111-D9S15-D9S110-qter. Recently, new centromeric markers have been reported. Analysis of these markers allowed us to localize the recombination breakpoint in some of the recombinant families. However, only one proximal recombination has been found with these markers. To increase the genetic information from FRDA families, we have analyzed the centromeric markers FR1, FR2, FR7, FR8, and FR5 in patients homozygous by descent. These were ascertained because parents were consanguineous or because they were homozygous for the entire haplotype D9S15 or D9S111-D9S5-D9S411E-D9S202. Haplotype divergence for, at least, two contiguous markers was observed in two patients homozygous for the core D9S111-FR2 haplotype and in one third-degree consanguineous family homozygous for haplotype D9S411E-FR5. Interpretation of divergence as the result of ancient meiotic crossovers allowed the definition of three new recombination events which place the FRDA locus within the interval defined by markers D9S411E and FR8. A consanguineous family with first-cousin parents showed homozygosity only at D9S202 and FR2. Further investigations are needed to discern whether two different mutations are segregating in the family or whether two recombinations, one distal and one proximal, have taken place.
Similar content being viewed by others


Introduction
Friedreich’s ataxia is an autosomal recessive neurodegenerative disease characterized by selective loss of large myelinated fibers in the peripheral nerves and by degeneration of the spinocerebellar and posterior tracts at the spinal cord. Clinical criteria for diagnosis include onset before the age of 25, progressive ataxia of gait and limbs, areflexia, and dysarthria [1]. Hypertrophic cardiomyopathy is often associated with the disease and an increased incidence of diabetes has been reported. A variable incidence of 1–4/100,000 has been recorded in the European population [2, 3]. The biochemical defect remains unknown and efforts addressed to understand the biological basis of the disease are based upon isolating the Friedreich’s ataxia gene (FRDA) by positional cloning. The FRDA locus was assigned to chromosome 9q13 by tight linkage with marker loci D9S15 [4] and D9S5 [5]. Linkage studies in several European and North American populations suggested that the FRDA locus lies within a 1-cM interval from D9S15 and D9S5 [6–8], which are 250 kb apart [9, 10]. Due to the tight linkage between markers and the disease locus, no centromeric-telomeric orientation of the linkage group could be established. Searches for recombination events have been initiated in large series, and recombinations between some marker loci and the FRDA locus were found in six families, suggesting the order cen-FRDA — D9S5 — D9S111 — D9S15 — D9S110 — qter as the most likely [11, 12]. Recently, Rodius et al. [13] developed a new generation of FRDA-linked microsatellite markers that map proximal to D9S5. Analysis of these markers in patients homozygous by descent and in association with founder haplotypes in isolated populations allowed the localization of three of the five markers centromeric to the FRDA locus, placing the disease locus in a cloned 450-kb interval [13].
We report here the search for haplotype recombinations in homozygous individuals by looking for loss of homozygosity of FRDA-associated marker loci in Spanish families. We have found one distal and two proximal ancient recombinations that support the hypothesis that the interval between D9S411E and FR8 markers is the critical region which contains the FRDA locus.
Subjects and Methods
Families
Fifty-two Spanish families with classical Friedreich’s ataxia were studied. Essential diagnostic criteria were confirmed in all patients, according to Harding [1]. In addition, all affected individuals underwent electrocardiogram and/or echocardiogram, and electrophysiological studies. A diagnosis of Friedreich’s ataxia was confirmed for the 7-year-old sister of an affected individual, because she demonstrated recent lower limb hyporeflexia. Diagnosis was based on the identification of the same D9S5/D9S15 haplotype found in the affected sister [14]. DNA samples were available from 76 affected and 64 unaffected children. In 18 families, two or more affected individuals could be sampled.
DNA Analysis
Genomic DNA was isolated from peripheral leukocytes using a standard phenol-chloroform extraction procedure. RFLP studies were performed by using dig-oxigenin-labelled probes, as described elsewere [15]. Microsatellite markers were detected by silver staining [16] of nondenaturing Polyacrylamide gels after PCR amplification according to the protocols given for each marker. RFLPs analyzed were MCTT12/MypI (D9S15) [17], and 26P/BstXI (D9S5) [7] and, in some cases DR47/TaqI [18] (D9S5). Microsatellite markers studied were MCT112/MS (D9S15) [7, 19], GS2 (D9S111) and GS4 (D9S110) [20], CA3 [21], FD1 (D9S411E) [22], MLS1 (D9S202) [23], FR1, FR2, FR5, FR7, and FR8 [13].
Results
A panel of 52 Spanish families, including 18 multiplex families, was analyzed for markers D9S15, D9S111, D9S5, D9S411E and D9S202, looking for phase-known recombinants. Two recombinant families, described elsewhere [11], were detected. Both families, LF1 and LF2, allowed the exclusion of the distal region to D9S111 for the FRDA gene location. The recombination breakpoint in family LF2 was detected between markers D9S5 and D9S15, whereas family LF1 was uninformative for all markers at D9S5, 26P and DR47, D9S411E, and D9S202. Subsequent analysis with the new proximal markers FR1 and FR5 was performed, and we could define the crossover breakpoint in family LF1 between D9S111 and FR1 in the paternal meiosis. Since FR1 is nonrecombinant, genetic analysis of this family supports the proximal location of the disease locus. No further known-phase recombination was observed in the remaining families.
In order to increase the genetic information from our series, we searched for ancient recombinations through the analysis of loss of homozygosity in consanguineous families and in patients homozygous for the linked haplotype (D9S15 or D9S111)-D9S5-D9S411E-D9S202. Eighteen such families were identified, amounting to 27 patients and 34 unaffected sibs. Consanguinity was documented in 6 families (parents were first cousins in 2 families and second cousins in 4 families). Patients from 12 families showed homozygosity for the (D9S15 or D9S111)-D9S202 haplotype.
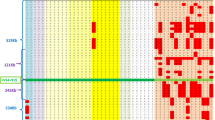
Five new markers, FR1, FR2, FR7, FR8, and FR5, were subsequently analyzed in these families, including patients, normal siblings, and parents when available. Figure 1 shows patients’ homozygosity or heterozygosity at the nine loci that we studied in the 18 families. Patients from consanguineous families and patients homozygous for haplotype D9S15 or D9S111 to D9S202 are represented in panel A and panel B, respectively. Figure 2 specifies each individual haplotype from the relevant families, in which loss of homozygosity was observed. Among the known consanguineous families, two had affected children with limited homozygosity (fig. 1, panel A). Individual LF38.5 was homozygous only for markers D9S202, FR1 and FR2 (fig. 2B) and individual LF64.3 was homozygous only for the proximal flanking marker FR5 and the distal flanking markers D9S5 and D9S411E (fig. 2D). Parents of patient LF38.5 were first cousins while parents of LF64.3 were second cousins. The possibility of two independent segregating mutations in both families must be taken into account. But regions of heterozygosity in individual LF38.5 might also reflect ancient recombinations from a grandparental haplotype, as discussed below. The four other known consanguineous families (LF2, LF16, LF46, and LF74) showed extensive evidence of homozygosity in the patients, in agreement with their parents cousinship. Nonrecombinant patients from family LF2 were homozygous for the interval between D9S15 (MCT112/MS polymorphism) and FR5 (Chamberlain et al. [11], and this study) except for the RFLP 26P/BstXI at D9S5 (fig. 2A). For more information, we analyzed the microsatellite CA3 that maps 20 kb proximal to D9S111 [21] and heterozygosity was observed again. As the parents are second cousins and two contiguous markers showed loss of homozygosity, this finding confirmed that the interval between CA3 and D9S5 does not contain the FRDA locus. Patients from family LF46 were homozygous for all markers tested but FR7 (fig. 2C). The same finding was observed in family LF36 (fig. 1, panel B), where the affected patient was homozygous for 7 markers, 6 of which were contiguous, but heterozygous at FR7 and D9S15. Whether these represent ancient recombination events, where the proximal FR5 locus gained the same allele as the one on the ancestral haplotype linked with the disease, or represent microsatellite mutations at FR7 by slippage, is not clear and needs further investigation.

Status of patients from each family for each locus, shown by shading of the appropriate square. Panel A = documented consanguineous families; panel B = families with patients homozygous for the core D9S15 or D9S111-D9S202 haplotype.

Pedigree and haplotypes of consanguineous families and families homozygous for the haplotype D9S15 or D9S111 to D9S202, for which allelic divergence at some markers has been observed.
More interesting, analysis of FR markers in patients homozygous for the haplotype (D9S15 or D9S111)-D9S5-D9S411E-D9S202 showed loss of homozygosity for at least two contiguous proximal markers in two families, in which FR1 and FR2 remained homozygous. Homozygosity by chance for D9S111 to FR2 is expected to occur with a frequency of 3 × 10−3 [13, 24], despite linkage disequilibrium between D9S5 and D9S411E and between MLS1 and FR1 at D9S202. It is therefore a good argument in favor of homozygosity by ancient nondocumented consanguinity, where divergence at two contiguous markers represents ancient recombinations, as slippage at the two or more microsatellites would be extremely unlikely. Patient LF49.4 (fig. 2E) was heterozygous at FR7 and FR5. FR8 remained homozygous, which suggests that the inferred recombination occurred between FR8 and FR7 and would place FR7 proximal to FR8. Divergence from homozygosity at FR8, FR7, and FR5 was observed in patient LF53.3 (fig. 2F). Taking both patients together, the region beyond FR8 towards the centromere was excluded as containing the FRDA locus. A third patient in family LF18 was heterozygous at FR5. Lack of more DNA samples prevented further genetic analysis in this family (fig. 2G). The patients from the other eight families with homozygosity for the haplotype (D9S15 or D9S111) to D9S202 were all homozygous for the FR markers, confirming homozygosity by descent in these families (probability for homozygosity by chance being about 2 × 10−4 in patient from family LF50, heterozygous at D9S111, and less than 10−4 in the other families).
Discussion
Since its mapping on chromosome 9 in 1988 [4], the search for the FRDA gene has been largely based on the positional cloning strategy. Taking the D9S15/D9S5 linkage group as a starting point, new polymorphic linked markers have been reported [20–23], and different genetic strategies have been proposed to orient the linkage group on chromosome 9q and to map the FRDA locus relative to D9S15/D9S5. Early linkage disequilibrium studies did not support a stronger allelic association of the FRDA locus with any loci [5, 25]. Later, by analyzing 140 French and Italian families, Sirugo et al. [24] obtained evidence of a stronger allelic association between FRDA and D9S5, which decreased towards D9S110. The finding of six recombinant families [11, 12], indicated that the FRDA locus might be located centromeric to D9S15/D9S5, the most probable loci order being cen-FRDA-D9S5-D9S111-D9S15-D9S110-qter. Proximal markers, FD1 (D9S411E) [22] and MLS1 (D9S202) [23], were isolated, but no recombinations of known phase were observed in the region. To obtain more genetic information from our panel of FRDA families, we started the search for ancient recombination events based on the homozygosity mapping strategy [26]. This approach is based on the fact that patients from consanguineous marriages will be preferentially homozygous by descent at the disease locus and, consequently, for an extended linked haplotype. Therefore, loss of haplotype homozygosity can be interpreted as the result of ancient recombinations. The same strategy can be applied to patients where homozygosity of linked haplotypes can prove unknown ancient consanguinity.
Very recently, a new set of proximal micro-satellite markers have been reported by two of us [13]. By typing a previously described recombinant family [12] with these markers, Rodius et al. [13] placed FR1 distal to FRDA. Moreover, analysis of haplotype divergence in patients homozygous by descent and in populations with a strong founder effect, allowed the definition of FR7/FR8 and FR5 as proximal flanking markers, and placed the FRDA locus in a 450-kb interval between FR1 and FR7/FR8 [13].
We analyzed FR1 and FR5 markers in our 52 FRDA families, looking for known-phase distal and proximal recombinations, respectively. None were found. We could place, however, the recombination breakpoint in family LF1 between FR1 and D9S111, indicating that the FRDA locus maps centromeric to D9S111. We then decided to scrutinize our FRDA families with known or suspected consanguinity for homozygosity with all the new centromeric markers FR1, FR2, FR8, FR7 and FR5. Eighteen families were analyzed, 6 with known consanguinity and 12 where homozygosity of markers D9S15 or D9S111 to D9S202 suggested an ancient relationship. In 11 families, homozygosity was found with all FR markers. Of the remaining seven families, six were also homozygous with FR1 and FR2, but had loose homozygosity at FR8 and/or FR7 and/or FR5. Several explanations may account for this loss of homozygosity: recombinations, microsatellite mutations (slippage), and different mutations sharing a limited haplotype by chance.
Heterozygosity observed with two and three contiguous markers in two families excludes the possibility of microsatellite mutations, as having multiple such events is very improbable. Homozygosity by chance is 6 × 10−4 and 3 × 10−3, respectively, and seems also unlikely. In fact, among more than 200 healthy individuals, none was found to be homozygous for the entire haplotype D9S15 or D9S111 to FR2. In both cases, loss of homozygosity seems, therefore, to be the result of ancient recombinations, placing the FRDA locus distal to FR7/FR8. In addition, loss of homozygosity at FR7-FR5 and not at FR8, in one of these families, would place FR8 distal to FR7, as the closest proximal FRDA flanking marker.
Loss of homozygosity at a single locus can be explained either by recombination or microsatellite mutation. Possible FR7 and FR5 mutations were described in founder haplotypes of isolated populations [13], with variations implying only one CA change. For the two FR7 divergences described here, slippage would imply a (CA)2 length variation.
Homozygosity limited to only a few markers in patients with known cousinship poses the problem of whether or not the disease is the consequence of consanguinity. In one family, parents were second cousins and homozygosity was restricted to FR5 and D9S411E-D9S5, both regions being rejected as containing the FRDA locus [13]. According to Lander and Botstein [26], the chance for not being homozygous by descent (i.e. being affected due to random meeting of disease alleles) is 0.22, assuming a disease allele frequency of 1/220 [2, 3], which is probably the case in this family. In the other family, parents were first cousins and the chance for not being homozygous by descent is only 0.06. An alternative explanation would be two recombinations, one proximal and one distal, placing the FRDA locus between D9S411E and FR8, as homozygosity was found at D9S202 (MLS1 and FR1) and FR2. The probability of two recombinations is low but, interestingly, this genomic region includes the actual FRDA candidate region. In addition, the homozygous patient haplotype MLS1-FR1-FR2 3-2-8 is in high linkage disequilibrium with the FRDA mutation in the Spanish population (p = 0.0002). This haplotype accounts for 37% (24/64) of FRDA chromosomes and 7% (4/56) of normal chromosomes [unpubl. data]. Both observations would support the two-recombinations explanation. On the other hand, false paternity is unlikely because there are two affected sibs. Lack of linkage to chromosome 9 is also unlikely, since no evidence of a second locus for Friedreich’s ataxia has been reported. Vitamin E deficiency linked to chromosome 8q [27, 28] was discarded because the patient had normal serum vitamin E values (15 µg/ml; normal range: 4–24 µg/ml). Nevertheless, further studies are required to solve this issue. This will be of interest, as the second possibility would corroborate the position of the FRDA locus in a small interval.
We have shown that investigating loss of homozygosity in FRDA patients homozygous by descent can be useful to find new recombination events. Application of this approach has improved the genetic information obtained from our FRDA families, increasing the number of informative recombinations between the FRDA locus and linked markers. These events would place the FRDA locus within the interval defined by D9S411E and FR8 markers, supporting the recent results by Rodius et al. [13]. Investigations with new markers mapping in that interval will allow us to narrow down the FRDA locus for positional cloning strategies.
References
Harding AE: Friedreich’s ataxia: A clinical and genetic study of 90 families with analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981;104:589–662
Winter RM, Harding AE, Baraitser M, Bravery MB: Intrafamilial correlation in Friedreich’s ataxia. Clin Genet 1981;20:419–427
Romeo G, Menozzi P, Ferlini A, Fadda S, DiDonato S, Uziel G, Lucci B, et al: Incidence of Friedreich ataxia in Italy estimated from consanguineous marriages. Am J Hum Genet 1983;35:523–529
Chamberlain S, Shaw J, Rowland S, Wallis J, South S, Nakamura Y, von Gabain A, et al: Mapping of the mutation causing Friedreich’s ataxia to chromosome 9. Nature 1988;334:248–250
Fujita R, Agid Y, Trouillas P, Seck A, Tommasi-Davenas C, Driesel AJ, Olek K, et al: Confirmation of linkage of Friedreich’s ataxia to chromosome 9 and identification of a new closely linked marker. Genomics 1989;4:110–111
Chamberlain S, Shaw J, Wallis J, Rowland A, Chow L, Farrall M, Keats B, et al: Genetic homogeneity at the Friedreich ataxia locus on chromosome 9. Am J Hum Genet 1989;44:518–521
Fujita R, Hanauer A, Sirugo G, Heiling R, Mandel JL: Additional polymorphisms at marker loci D9S5 and D9S15 generate extended haplotypes in linkage disequilibrium with Friedreich ataxia. Proc Natl Acad Sci USA 1990;87:1776–1800
Pandolfo M, Sirugo G, Antonelli A, Weitnauer L, Ferretti L, Leone M, Dones I, et al: Friedreich ataxia in Italian families: Genetic homogeneity and linkage disequilibrium with the marker loci D9S5 and D9S15. Am J Hum Genet 1990;47:228–235
Fujita R, Hanauer A, Vincent A, Mandel JL, Koenig M: Physical mapping of two loci (D9S5 and D9S15) thightly linked to the Friedreich’s ataxia locus (FRDA) and identification of nearby CpG islands by pulse-field gel electrophoresis. Genomics 1991;10:915–920
Fujita R, Sirugo G, Duclos F, Abderrahim H, Le Pasleir D, Cohen D, Brownstein BH, et al: A 530 kb YAC contig tightly linked to Friedreich ataxia locus contains 5 CpG clusters and a new highly polymorphic microsatellite. Hum Genet 1992;89:531–538
Chamberlain S, Farrall M, Shaw J, Wilkes D, Carvajal J, Hillerman R, Doudney K, et al: Genetic recombination events which position the Friedreich ataxia locus proximal to the D9S15/D9S5 linkage group on chromosome 9q. Am J Hum Genet 1993;52:99–109
Belal S, Kyproula P, Sirugo G, Ben Hamida C, Panos I, Hentati F, Beckman J, et al: Study of large inbred Friedreich ataxia families reveals a recombination between D9S15 and the disease locus. Am J Hum Genet 1992;51:1372–1376
Rodius F, Duclos F, Wrogemann K, Le Paslier D, Ougen P, Billault A, Belal S, et al: Recombinations in individuals homozygous by descent localize the Friedreich ataxia locus in a cloned 450-kb interval. Am J Hum Genet 1994;54:1050–1059
Palau F, Monrós E, Prieto F, Vilchez JJ, Lopez-Arlandis JM: Genetic diagnosis of Friedreich ataxia. Lancet 1991;338:1087.
Palau F, Löfgren A, De Jonghe P, Bort S, Nelis E, Sevilla T, Martin JJ, Vilchez J, Prieto F, Van Broeckhoven C: Origin of the de novo cuplication in Charcot-Marie-Tooth disease type 1A: Unequal nonsister chromatid exchange during spermatogenesis. Hum Mol Genet 1993;2:2031–2035
Beidler JL, Hilliard PR, Rill RL: Ultrasensitive staining for nucleic acids with silver. Anal Biochem 1982;126:374–380
Carlsson M, Nakamura Y, Krapcho K, Fujimoto E, O’Connell P, Leppert M, Lathrop GM, et al: Isolation and mapping of a polymorphic DNA sequence pMCT112 on chromosome 9q (D9S15). Nucleic Acids Res 1987;15:10614.
Orzechowski HDD, Henning J, Winter P, Grzeschik KH, Olek K, Driesel AJ: A human single-copy DNA probe (DR47) detects a TaqI RFLP on chromosome 9 (D9S5). Nucleic Acids Res 1987;15:6310.
Wallis J, Williamson R, Chamberlain S: Identification of a hypervariable microsatellite polymorphism within D9S15 tightly linked to Friedreich’s ataxia. Hum Genet 1990;85:98–100
Sirugo G, Keats B, Fujita R, Duclos F, Purohit K, Koenig M, Mandel JL: Friedreich ataxia in Lousiana-Acadians: Demonstration of a founder effect by analysis of microsatellite generated extended haplotypes. Am J Hum Genet 1992;50:559–566
Pugachev VV, Polyahov L, Prokunina L, Evgrafov OV: Molecular and genetic study of FRDA locus in Russia. Int Workshop on the Molecular Genetics of Friedreich’s and Dominant Ataxias, Capri 1993, pp 18–19.
Duclos F, Boschert U, Sirugo G, Mandel JL, Koenig M: New gene in the region of the Friedreich ataxia locus encodes a putative transmembrane protein expressed in the nervous system. Proc Natl Acad Sci USA 1993;90:109–113
Pandolfo M, Murano M, Cocozza S, Redolfi EM, Pianese L, Cavalcanti F, Monticelli A, et al: A dinucleotide repeat polymorphism (D9S202) in the Friedreich’s ataxia region on chromosome 9q13-q21.1. Hum Mol Genet 1993;2:822.
Sirugo G, Cocozza S, Brice A, Cavalcanti F, De Michele G, Dones I, Filla A, et al: Linkage disequilibrium analysis of Friedreich’s ataxia in 140 Caucasian families: Positioning of the disease locus and evaluation of allelic heterogeneity. Eur J Hum Genet 1993;1:133–143
Hanauer A, Chery M, Fujita R, Driesel AJ, Gilgenkranz S, Mandel JL: The Friedreich ataxia gene is assigned to chromosome 9q13-q21 by mapping tightly linked markers and shows linkage disequilibrium with D9S15. Am J Hum Genet 1990;46:133–137
Lander ES, Botstein D: Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science 1987,236:1567–1570.
Ben Hamida M, Belal S, Sirugo G, Ben Hamida C, Panayides K, Ioannou P, Beckmann J, et al: Friedreich’s ataxia phenotype not linked to chromosome 9 and associated to selective autosomal recessive vitamin E deficiency in two inbred Tunisian families. Neurology 1993;43:2179–2183
Ben Hamida C, Doerflinger N, Belal S, Linder C, Retenauer L, Dib C, Gyapay G, et al: Localization of Friedreich ataxia phenotype associated with selective vitamin E deficiency to proximal 8q by homozygosity mapping. Nat Genet 1993;5:195–200
Acknowledgements
We thank Dr. Y. Nakamura and Dr. J.L. Mandel for probes MCT112 and 26P, respectively, and the Friedreich’s ataxia families and the Asociación Española de Ataxias Hereditarias for their enthusiastic cooperation. This study was supported by grants from the Comisión Interministerial de Ciencia y Tecnología, CICYT and from the Generalitat Valenciana (FP), from the Association Française contre les Myopathies and the Groupement de Recherche et d’Etudes sur les Génomes (MK) and from Italian Telethon (MP). EM and JC are recipients of a Generalitat Valenciana Fellowship.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Monrós, E., Smeyers, P., Rodius, F. et al. Mapping of Friedreich’s Ataxia Locus by Identification of Recombination Events in Patients Homozygous by Descent. Eur J Hum Genet 2, 291–299 (1994). https://doi.org/10.1159/000472373
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472373
Key Words
This article is cited by
-
A family segregating a Friedreich ataxia phenotype that is not linked to the FRDA locus
Human Genetics (1996)
