
Abstract
Alagille syndrome (AGS) has been assigned to 20p11.23-20p12.2 according to minimum overlap between deletions observed on the chromosome 20 short arm of 9 patients. We report here the localisation of 5 microsatellite markers (D20S41, D20S48, D20S50, D20S56, and D20S58) within the deletion of one AGS patient. This study allows an estimation of the genetic extent of this deletion as being between 30 and 36 cM, and demonstrates its paternal origin. The search for submicroscopic deletions in 23 AGS patients, by typing these 5 markers, failed to reveal allelic loss. However, these results lead to the proposition that the AGS locus lies in one of the seven intervals defined by the six microsatellite markers in the region flanked by D20S5 and D20S18.
Similar content being viewed by others


Introduction
Alagille syndrome (AGS; MIM 118450) is a complex malformative disorder characterised by five main features: a paucity of intrahepatic bile ducts, peripheral pulmonary artery stenosis, butterfly-like vertebrae, posterior embryotoxon, and peculiar facies [1]. This disorder, inherited as an autosomal dominant trait with quasi-complete penetrance [2], has been assigned to 20p11.23-20p12.2 by a series of observations of AGS patients with 20p deletions (fig. 1) [see ref. 3 for a review].

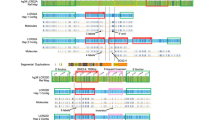
Genetic and physical map of the AGS region of the short arm of chromosome 20 and a schematic representation of the smallest region of overlap (SRO) on a chromosome 20 R-banded idiogram. A The SRO was defined on cytogenetics grounds according to previous reports of 9 deletions. The deletions are numbered from 1 to 9 [for del 1–5,8 and 9, ref. 3; for del 6, ref. 5; for del 7, ref. 11]. B The genetic map is connected to a chromosome 20 idiogram by the physical localisation of the RFLP markers. This map, established according to a previous report [4], is scaled in centimorgans (cM) using the Haldane mapping function. The markers located outside the deletion studied (del 6) are boxed.
The hypothesis that AGS is due to deletion involving contiguous genes would account for the variable phenotypic expression and the complexity of this disorder [5–7]. This led to the suggestion that cytogenetically normal patients have a submicroscopic deletion [6], as has been demonstrated for deletion-related diseases such as the DiGeorge syndrome [8], the WAGR contiguous gene syndrome [9], and also in the monogenic Miller-Dieker syndrome [10]. The search for submicroscopic deletions is a valuable approach to circumscribe the AGS region.
The 20p11.23-20p12.3 deletion detected in one of our AGS patients [5], one of the smallest deletions observed, can be used to characterise the smallest region of overlap (SRO) of the cytogenetically described deletions (fig. 1). Three RFLP markers (D20S5, D20S6, and D20S18) map within this deletion [5]. No deletions were found in AGS patients analysed with fluorescent in situ hybridisation of the cosmid probe D20S6 [6]. Taking into account the conservation of D20S5 and D20S18 in the DNA of AGS patients presenting 20p deletions, they are therefore not adequate markers for further screening [11; I. Hansmann, pers. commun.].
Microsatellites are informative tools to search for allelic loss in genotyping studies. We used nine microsatellites, D20S40, D20S41, D20S42, D20S45, D20S48, D20S50, D20S56, D20S58, and D20S59, mapped on the chromosome 20 short arm [4] and spanning the region flanked by D20S5 and D20S18 (fig. 1), in a genotyping study in the families of 23 patients.
Material and Methods
Patients and Families
Clinical data of the deleted proband and his family (AGSN1) have been published previously [5]. Among the 22 other families studied, 21 were ascertained through an affected child and one (AGSG2) via two affected children. Clinical data on the 23 probands and family investigations were reported previously [2]. All patients presented with at least four main features including paucity or stenosis. All the patients had a chromosome analysis; none of them exhibited 20p deletions.
Genotyping
Allele determination was performed by the polymerase chain reaction using primers described previously [4]; electrophoresis, transfer and hybridisation were according to Hazan et al. [12].
Results
Determination of the Genotypes in the Family of the Deleted Proband
The nine microsatellites were localised on a genetic map with reference to D20S5, D20S6, and D20S18 (table 1). However, the RFLP markers were not precisely mapped (support interval of the maximum likelihood location 100:1) [4]. These 9 markers were typed in the AGSN1 family (table 1) in order to precisely localise them with respect to the deletion. D20S41, D20S58, and D20S48 were deleted as shown by the lack of paternal alleles in the proband. The genotypes of D20S59, D20S42, and D20S45 were heterozygous in the proband, indicating that they map outside the deletion (fig. 2). The genotypes of D20S56 and D20S50 in the proband could be interpreted as being either homo- or hemizygous. D20S56 and D20S50, mapped between D20S41 and D20S48 [4], were therefore assumed to be deleted. The situation of D20S40 remains unclear, as it is not easy to determine allele dose by this technique and, therefore, a homozygous individual cannot be distinguished from a hemizygote.

Autoradiogram illustrating two microsatellites, D20S58 (a) and D20S59 (b) amplified in the family AGSN1. The numbers below the autoradiograms represent the genotype of the individuals. The loss of the paternal allele 1 in the deleted proband indicates that D20S58 maps within the deletion. The heterozygote genotype obtained with D20S59 in the deleted proband (b) confirms its position outside the deletion.
This study provided physical data which allowed a precise genetic mapping of the AGS region. The retention of D20S42 and the deletion of D20S18 allowed us to map D20S18 distal to D20S42. From these results, the maximal and the minimal genetic extents of this deletion can be determined as 36cM between D20S59 and D20S42 and 30 cM between D20S5 and D20S18, respectively. Finally, these results demonstrated the paternal origin of the deletion.
Screening of Microdeletions
Each of the 67 individuals from the 22 families was typed with the 9 microsatellite markers. The genotypes of the 23 AGS patients were either heterozygous or compatible with a homozygous status. Thus, because of the unreliability of allelic dosage, this analysis failed to reveal allelic loss. None of the five markers mapped within the deletion of our AGS patient (D20S41, D20S48, D20S50, D20S56, and D20S58) were homozygous in all the other AGS patients. The heterozygous genotype of these five markers allowed their exclusion from a submicroscopic deletion in at least 50% of AGS patients (table 2).
Discussion
Only three molecular studies of AGS have been reported [5, 6, 11]. We report here the molecular study of an AGS deletion using 9 microsatellite markers and three RFLP markers, to genetically map it more precisely. D20S59 and D20S42 are the distal and proximal non-deleted extremities, respectively. D20S5, D20S6, D20S18, D20S41, D20S48, D20S50, D20S56, and D20S58 are located within the deleted region. Taking into account the exclusion of D20S5 and D20S18 from inside the SRO, its maximal genetic extent can be estimated to be 30 cM between these two markers.
None of the typed microsatellites was deleted from the DNA of all the AGS patients, indicating that none of these markers was part of the minimal deletion causing AGS. Presumably, the minimal deletion maps between two of these microsatellites. These results allowed us to propose 7 candidate intervals (defined by the six microsatellite markers in the region flanked by D20S5 and D20S18) for the mapping of the AGS locus. D20S6, previously designated a ‘reference marker’ for chromosome 20, was no longer considered because of observed intralocus recombination [13].
Several other microsatellites have recently been located on the short arm of chromosome 20 [14]. The mapping of these markers, with respect to those we studied, will facilitate the identification of the AGS locus.
References
Alagile D, Estrada A, Hadchouel M, Gautier M, Odièvre M, Donrmergues JP: Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): Review of 80 cases. J Pediatr 1987;110:195–200
Dhorne-Pollet S, Deleuze JF, Hadchouel M, Bonaïti-Pellié C: Segregation analysis of Alagille syndrome. J Med Genet 1994;31:453–457
Anad F, Burn J, Matthews D, Cross I, Davison BCC, Mueller R, Sands M, Lillington DM, Eastham E: Alagille syndrome and deletion of 20p. J Med Genet 1990;27:729–737
Hazan J, Dubay C, Pankowiak MP, Becuwe N, Weissenbach J: A genetic linkage map of human chromosome 20 composed entirely of microsatellite markers. Genomics 1992;12:183–189
Zhang FR, Deleuze JF, Aurias A, Dutrillaux AM, Hugon RN, Alagille D, Thomas G, Hadchouel M: Interstitial deletion of the short arm of chromosome 20 in arteriohepatic dysplasia (Alagille syndrome). J Pediatr 1990.116:73–77.
Desmaze C, Deleuze JF, Dutrillaux AM, Thomas G, Hadchouel M, Aurias A: Screening of microdeletions of chromosome 20 in patients with Alagille syndrome. J Med Genet 1992;29:233–235
Schmickel RD: Contiguous gene syndromes: A component of recognisable syndromes. J Pediatr 1986;109:231–241
Scambler PJ, Carey AH, Wyse RKH, Roach S, Dumanski JP, Nordenskjold M, Williamson R: Microdeletions within 22q11 associated with sporadic and familial Di-George syndrome. Genomics 1991;10:201–206
Fantes JA, Bickmore WA, Fletcher JM, Ballesta F, Hanson IM, van Heyningen V: Submicroscopic deletions at the WAGR locus, revealed by non radioactive in situ hybridisation. Am J Hum Genet 1992;51:1286–1294
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH: Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature 1993;364:717–721
Schnittger S, Höfers C, Heidemann P, Beermann F, Hansmann I: Molecular and cytogenetic analysis of an interstitial 20p deletion associated with syndromic intrahepatic ductular hypoplasia (Alagille syndrome). Hum Genet 1989;83:239–244
Hazan J, Lamy C, Melki J, Munnich A, de Recondo J, Weissenbach J: Autosomal dominant familial spastic paraplegia is genetically heterogeneous and one locus maps to chromosome 14q. Nature Genet 1993;5:163–167
NIH/CEPH Collaborative Mapping Group: A comprehensive genetic linkage map of the human genome. Science 1992;258:67–86
Weissenbach J, Gyapay G, Dib C, Vignal A, Morissette J, Milasseau P, Vaysseix G, Lathrop M: A secondgeneration linkage map of the human genome. Nature 1992,359: 794–801.
Acknowledgements
This work was supported by grants from Association Française contre les myopathies (AFM) and Groupement de Recherches et d’Etudes sur les Génomes (GREG). J.F.D. and J.H. were supported by grants from AFM. S.D. was supported by grants from Ministère de l’Enseignement Supérieur et de la Recherche (MESR).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Deleuze, JF., Hazan, J., Dhorne, S. et al. Mapping of Microsatellite Markers in the Alagille Region and Screening of Microdeletions by Genotyping 23 Patients. Eur J Hum Genet 2, 185–190 (1994). https://doi.org/10.1159/000472362
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472362
Key Words
This article is cited by
-
Mutations in the human Jagged1 gene are responsible for Alagille syndrome
Nature Genetics (1997)
-
Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1
Nature Genetics (1997)
-
Endokrinologische und metabolische Komplikationen des Alagille-Syndroms
Medizinische Klinik (1997)
-
Fatty acid content in lymphocytes from children with syndromic paucity of interlobular bile ducts, Alagille syndrome
Journal of Inherited Metabolic Disease (1995)
