
Abstract
Schizophrenia (SZ) is a neurodevelopmental disorder with a broad symptomatology, including cognitive symptoms that are thought to arise from the prefrontal cortex (PFC). The neurobiological aetiology of these symptoms remains elusive, yet both impaired redox control and PFC dysconnectivity have been recently implicated. PFC dysconnectivity has been linked to white matter, oligodendrocyte (OL) and myelin abnormalities in SZ patients. Myelin is produced by mature OLs, and OL precursor cells (OPCs) are exceptionally susceptible to oxidative stress. Here we propose a hypothesis for the aetiology of cognitive symptomatology in SZ: the redox-induced prefrontal OPC-dysfunctioning hypothesis. We pose that the combination of genetic and environmental factors causes oxidative stress marked by a build-up of reactive oxygen species that, during late adolescence, impair OPC signal transduction processes that are necessary for OPC proliferation and differentiation, and involve AMP-activated protein kinase, Akt-mTOR-P70S6K and peroxisome proliferator receptor alpha signalling. OPC dysfunctioning coincides with the relatively late onset of PFC myelination, causing hypomyelination and disruption of connectivity in this brain area. The resulting cognitive deficits arise in parallel with SZ onset. Hence, our hypothesis provides a novel neurobiological framework for the aetiology of SZ cognitive symptoms. Future research addressing our hypothesis could have important implications for the development of new (combined) antioxidant- and promyelination-based strategies to treat the cognitive symptoms in SZ.
Similar content being viewed by others


Introduction
Schizophrenia (SZ) is a neurodevelopmental disorder with positive, negative and cognitive symptoms. Current treatments only target positive symptoms, therefore identifying new treatment strategies that aim at negative and cognitive symptoms is of crucial importance. To achieve this, the elucidation of the neurobiological correlates underlying these symptoms is a necessary first step. Cognitive symptoms of SZ, the focus of this review, include poor executive functioning and are thought to arise from the prefrontal cortex (PFC).1, 2 Both redox imbalance and PFC dysconnectivity have been implicated in the aetiology of these symptoms.
SZ is associated with redox imbalance
Redox imbalance is a state of high oxidative stress caused by an imbalance between the production of reactive oxygen species (ROS) and antioxidants that reduce ROS. A continuous balance between ROS production and reduction is crucial to maintain ROS-dependent cellular processes as well as to prevent ROS-induced cell damage.
Environmental insults that are associated with SZ cause oxidative stress
One of the most important risk factors for the development of SZ is the activation of the maternal immune system.3, 4 The mechanism by which maternal immune activation affects brain development likely involves oxidative stress.5 For example, lipopolysaccharide (LPS) exposure during pregnancy induces the release of pro-inflammatory cytokines that induce ROS generation and peroxisomal dysfunction, whereas antioxidants such as N-acetyl cysteine can reverse the negative effects of LPS exposure on brain development.6 Other environmental factors associated with redox imbalance and SZ are prenatal malnutrition and maternal stress during pregnancy.7, 8, 9, 10, 11, 12 For example, low protein intake during pregnancy has been shown to induce mitochondrial dysfunction and a decrease in endogenous antioxidants, resulting in higher ROS production.13 In addition, obstetric events, such as hypoxia, and environmental insults later in life, such as social stress, are associated with oxidative stress and represent risk factors for SZ.14, 15, 16, 17, 18, 19, 20
Redox imbalance in SZ patients
Genetic studies have shown associations between oxidative stress gene polymorphisms and SZ,21, 22 including genetic variations in glutathione cysteine ligase (GCL) and several glutathione-S-transferases,23, 24, 25 both involved in the synthesis of the endogenous antioxidant glutathione. Fibroblasts of patients carrying genetic variations in GCL display lower glutathione and GCL protein expression, and thus redox imbalance.25 Unlike genetic association studies, the available genome-wide association studies (GWASs) have not provided convincing evidence for oxidative stress-related genetic predisposition in SZ, and therefore additional GWASs with larger sample sizes may be necessary.
In addition, both downregulation of components of the antioxidant synthesis pathway and increases in ROS levels have been observed in SZ patients. For instance, total antioxidant and glutathione plasma levels are lower in non-medicated, medicated, first-episode as well as chronic SZ patients,26, 27, 28, 29 in line with the reduced glutathione levels found in the PFC and cerebral spinal fluid of SZ patients30, 31 and in post mortem SZ brains,32 in which abnormal redox-related protein expression has also been found.33 Furthermore, peripheral levels of ROS are increased, and those of glutathione peroxidase and superoxide dismutase are decreased in SZ patients,34, 35, 36, 37, 38, 39 independent of drug use or disease stage. Hence, both lower levels of antioxidants and higher levels of ROS are core features of the disorder and are not influenced by disease progression or medication use, indicating that redox imbalance is a primary characteristic of the disorder. Interestingly, in SZ patients, deficits in executive functioning are correlated with higher ROS levels and lower antioxidant-related protein levels,38 directly linking redox imbalance to cognitive dysfunction.35
Redox imbalance in SZ rodent models
The N-methyl-D-aspartate-antagonist MK-801-induced rat model for SZ shows increased oxidative stress specifically in the PFC,40 although higher levels of brain mitochondrial ROS have been found in a ketamine-induced rat model.41 Inversely, glutathione depletion in rats leads to SZ-like phenotypes.42, 43, 44 In addition, knockout mice that lack a crucial subunit of the GCL enzyme show significant reduction of glutathione levels in the anterior cortex,45 especially during the prepuberal period, which is followed by SZ-like behaviour in the time frame of disease onset46 and SZ-like neural changes in the hippocampus (HIP) of the adult knockout mice, including an increase in oxidative stress and a decrease in the number of parvalbumin (PV) interneurons.47 Therefore, redox imbalance may represent the main trigger for brain alterations before disease onset, which negatively influence cognition later on.
Prefrontal dysconnectivity is associated with cognitive symptoms of SZ
Diffusion magnetic resonance imaging reveals alterations in white matter (WM) integrity, that is, lower fractional anisotropy (FA; for a review, see Wheeler and Voineskos48), in both medicated and non-medicated SZ subjects.49, 50 Importantly, even before SZ disease onset, a reduced WM integrity occurs in frontal areas and advances in further stages of the disorder to more caudal and posterior regions.50, 51, 52, 53, 54, 55
WM abnormalities in SZ are associated with cognitive symptomatology
Correlations between cognition and frontal WM integrity have been reported in healthy individuals.56, 57 In chronic SZ, abnormalities in cognitive processing speed are associated with WM disruptions in, among others, frontal areas,58, 59, 60 and in first-episode SZ patients a lower frontal WM integrity is correlated with more severe cognitive symptoms.61, 62 Interestingly, deficit SZ (that is, SZ with strong cognitive impairment63, 64) is associated with severe WM abnormalities.65, 66, 67, 68 Furthermore, cognitive symptomatology of SZ patients worsens as the disease progresses, in line with the ongoing WM alterations.69, 70
Origin of lower FA in SZ PFC
A low FA value in diffusion magnetic resonance imaging is indicative of alterations in WM that can be attributed to several cellular factors, including reduced myelination and aberrant axonal properties.71 Diffusion tensor as well as kurtosis imaging reveal a lower FA and increased radial diffusivity in combination with no changes in axial diffusivity in the frontal lobe of SZ patients.72 This indicates that myelin rather than axonal abnormalities form the neurobiological basis of the diffusion magnetic resonance imaging aberrations in SZ. Other diffusion studies show similar results.73, 74 Direct evidence for axonal degeneration in SZ is indeed lacking. Furthermore, magnetisation transfer ratio, a more specific imaging measure for myelin, shows lower myelin levels in, among others, the PFC of SZ patients compared with controls.75, 76 These low myelin levels predict impaired processing speed in SZ and link decreased myelination to cognitive symptoms of the disorder.77, 78
SZ is associated with oligodendrocyte abnormalities and decreased myelination
Myelin is produced by oligodendrocytes (OLs) that are derived from OL precursor cells (OPCs) in the developing as well as the adult brain.79, 80, 81 Plasticity in the formation and retraction of myelin sheaths by OLs also occurs from early childhood to adulthood.80, 81 Neuronal activity can instruct OPCs to divide and mature, and can stimulate myelin sheath production by OLs,82 leading to increased myelination and improved behavioural performance.83 Conversely, reduced neuronal stimulation by social isolation impairs myelination, which correlates with behavioural and cognitive dysfunction.84, 85 Accordingly, altered myelination dynamics may have a major role in cognition as well as in psychiatric disorders like SZ.
Evidence from human post mortem studies
In the PFC of SZ patients, lower OL size and regional-specific differences in OL density alongside higher levels of OL apoptosis and necrosis have been observed, accompanied by lower levels of myelin.86, 87, 88, 89, 90 Furthermore, expression of the myelin-associated proteins 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase and myelin-associated glycoprotein is significantly lower in SZ anterior frontal cortex,90 and differential mRNA expression of these two and other myelin-related genes has been observed in SZ dorsolateral PFC.91 Overall, there is abundant evidence for an OL as well as a myelin deficit in the PFC of SZ post mortem brain.
Evidence from rodent models
Evidence for a myelin deficit in SZ is also provided by studies on rodent models that range from pharmacological and transgenic to neurodevelopmental models. For example, administration of the MK-801 in adulthood is used as a model for SZ (for a review see Neillet al.92) and alters brain expression of, among others, platelet-derived growth factor (PDGF), proteolipid protein, myelin basic protein and 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase,93 and decreases WM volume, together with myelin sheath degeneration.94 Furthermore, mice transgenic for SZ-associated locus G72/G30 show SZ-like behavioural traits and myelin-related protein expression changes.95 In addition, severe hypomyelination has been observed in mice mutant for the myelination-associated gene quaking (a gene downregulated in SZ) alongside structural abnormalities of myelin sheath thickness and composition.96, 97 Moreover, rodent models for demyelination display SZ-like behavioural abnormalities, for example, cuprizone demyelination leads to reduced expression of several OL-related transcripts and diminished ability to perform a SZ-relevant cognitive flexibility task.98
Genetic evidence
OL-related gene variants correlate with reduced WM integrity and cognitive performance.99, 100 Nevertheless, candidate gene association studies and a large meta-analysis of genetic risk for SZ have shown that myelin- and OL-related genes are not significantly associated with the disorder.101, 102, 103, 104, 105 Therefore, in most cases the myelin pathology observed in SZ likely reflects a secondary phenotype with an indirect, non-genetic cause.
Redox imbalance can cause an OPC maturation deficit
OPCs and OLs contain exceptionally high amounts of ROS (six times as much), three times lower glutathione concentration and 20-fold higher free-iron levels than astrocytes,106, 107 probably because their myelin synthesis entails a high metabolic rate.108 This means that OPCs and OLs are constantly under a high degree of oxidative stress to which the cells are already more susceptible. In fact, redox changes of only 15–20% can already influence signal transduction pathways such as PDGFα stimulation of OPC proliferation and maturation.109 The susceptibility of OPCs and OLs to oxidative stress has serious implications for the process of myelination. For instance, oxidative stress leads to downregulation of myelin-related gene expression in human OLs in vitro,110 and reduced myelin basic protein expression and OL number in the rat brain.111, 112, 113 Hence, the myelination abnormalities observed in SZ may well be due to oxidative stress-related OPC dysfunctioning.
Hypothesis of redox-induced prefrontal OPC dysfunctioning
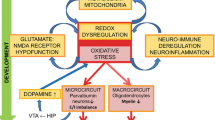
On the basis of the above, we here propose the redox-induced prefrontal OPC-dysfunctioning hypothesis of cognitive symptomatology in SZ. This hypothesis states that in SZ the combination of environmental factors and genetic predisposition causes oxidative stress, marked by a build-up of ROS in OPCs (Figure 1). During late adolescence, the high ROS levels impair OPC signal transduction processes that are necessary for their proliferation and differentiation. OPC dysfunctioning coincides with the relatively late onset of PFC myelination, and causes hypomyelination and disruption of connectivity in this brain area. The resulting cognitive symptoms coincide with SZ onset.

Flowchart of the redox-induced prefrontal OPC-dysfunctioning hypothesis. Environmental and genetic factors lead to a faulty antioxidant system, as well as redox imbalance resulting in OPC/OL proliferation and maturation arrest during adolescence, causing hypomyelination of the PFC, insufficient PFC functioning and subsequently the cognitive symptoms observed in SZ. OL, oligodendrocyte; OPC, OL precursor cell; PFC, prefrontal cortex; SZ, schizophrenia.
In the next sections, evidence for this hypothesis will be presented. First, the relationship between redox imbalance, hypomyelination and cognitive functioning in the PFC will be highlighted. Second, the molecular mechanisms underlying the impairment of OPC functioning by ROS will be discussed. Third, we will consider the critical developmental time period of PFC myelination and, in particular, of PFC hypomyelination in SZ.
ROS can cause OPC dysfunctioning
Baseline levels of oxidative stress in OPCs are high. In SZ, oxidative stress levels in OPCs are even higher because of extra ROS production by environmental factors as well as intracellular abnormalities that lead to extra ROS production and less ROS clearance (see above). The cause of OPC dysfunction in SZ may be explained by two different, but related, cellular pathways described below. In both pathways, ROS inactivates protein synthesis that is necessary for OPC proliferation and differentiation via the mammalian target of rapamycin (mTOR)-P70S6K pathway. The inactivation of the latter pathway leads to OPC proliferation arrest, apoptosis and hypomyelination.114
Figure 2 presents a molecular map that is based on the literature described below and depicts the interactions among various molecules inactivating the mTOR-P70S6K pathway in SZ OPCs.

Molecular map of pathways that lead ROS to cause OPC dysfunctioning in SZ. A molecular map is essential to elucidate the neurobiological mechanisms underlying the impairment of OPC functioning by ROS in the SZ PFC. The map shows that two cellular pathways result in a reduced activation of the mTOR-P70S6K pathway under conditions of increased ROS production and decreased antioxidant levels. The first pathway involves ROS-induced downregulation of PDGFRα, leading to sub-activation of the mTOR-P70S6K pathway. The second AMPK-related pathway leads to inhibition of the mTOR-P70S6K signalling cascade, as well as to downregulation of the transcription of proliferation- and differentiation-related genes. Inactivation of mTOR-P70S6K causes decreased protein synthesis for proliferation and differentiation, and consequently leads to OPC dysfunction. See section 'ROS can cause OPC dysfunctioning - Inactivation of the mTOR-P7056 pathway in SZ OPCs' for a description, Supplementary Table 1 for the pertinent references and the list of abbreviations for full names of all components of the molecular map. AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin; OPC, oligodendrocyte precursor cell; PDGF, platelet-derived growth factor; PFC, prefrontal cortex; ROS, reactive oxygen species; SZ, schizophrenia.
Inactivation of the mTOR-P70S6 pathway in SZ OPCs
The relatively active metabolism in OPC mitochondria leads to the production of ROS as a by-product of the respiratory chain (Figure 2). The elevated ROS levels cannot be effectively reduced by glutathione because in OPCs glutathione levels are low. Excess ROS leads to an overstimulation of AMP-activated protein kinase (AMPK), which activates the tuberous sclerosis 1/2 complex. This complex prevents the activation of the mTOR-P70S6K pathway through inhibition of ras homologue enriched in brain (RHEB).115 Moreover, RHEB mediation of mTOR activity is necessary for OPC differentiation into myelinating OLs.116 In addition, AMPK stimulation causes enhanced biosynthesis of mitochondria via peroxisome proliferator-activated receptor gamma coactivator-1 alpha as well as the upregulation of glutathione and other antioxidants. However, these antioxidant levels are not sufficient to rescue the redox imbalance in SZ OPCs.117 Proliferator-activated receptor gamma coactivator-1 alpha transactivation of peroxisome proliferator receptor alpha inhibits transcriptional nuclear factor kappa-light-chain-enhancer of activated B cells,118 preventing efficient transcriptional activation of its target genes, thus contributing to OPC dysfunctioning.118, 119, 120 In addition, environmental factors implicated in SZ (for example, prenatal stress and malnutrition) may cause the production of cytokines such as tumour necrosis factor alpha.121, 122 This cytokine can activate AMPK, but also directly leads to both the mitochondrial uptake of calcium that might trigger apoptosis and to the additional activation of complex I of the respiratory chain, followed by an increase in ROS production.115
The other cellular pathway that can give rise to reduced activity of the mTOR-P70S6K pathway includes signalling via PDGFRα. As stated above, activation of this receptor is necessary for proliferation and maturation of OPCs. The increased levels of ROS in SZ OPCs cause stimulation of Fyn kinase, which in turn activates C-Casitas B-lineage Lymphoma.109, 123 This overactivation of C-Casitas B-lineage Lymphoma has been shown to decrease PDGFRα receptor numbers on the OPC cell membrane, reduce mTOR-P70S6K pathway activation and lower protein synthesis rate for proliferation and differentiation, disrupting OPC cell function.109, 124, 125 Interestingly, glutathione depletion, both in vivo and in vitro, inhibits Fyn-dependent maturation of OPCs, accompanied by reduced myelination.126
Proof of concept for the hypothesis that hypoactivation of the mTOR-P70S6K pathway leads to inhibition of OPC proliferation and maturation, and subsequently hypomyelination is provided by the fact that conditional mTOR knockout in mouse OPCs leads to various myelination defects.127 Furthermore, a number of studies have demonstrated that ERK1/2 signalling (which inhibits the tuberous sclerosis 1/2 complex and, therefore, increases mTOR-P70S6K signalling) can enhance myelination. For example, ERK1/2 signalling is implicated in the mechanism of action of diosgenin, a drug that enhances OPC differentiation and myelination,128 and of miconazole, which promotes remyelination in vitro and in animal models of multiple sclerosis (MS).129
In sum, a correct regulation of the AMPK, mTOR-P70S6K and ERK1/2 pathways is essential for OPC functioning and myelination. In SZ, these pathways are affected by increased oxidative stress, leading to OPC dysfunctioning and subsequently hypomyelination.
Redox imbalance, aberrant myelination and cognitive functioning are directly related
Evidence from SZ patients and rodent models
Low glutathione levels are correlated with reduced WM integrity in the medial PFC of SZ patients and in PFC myelin of GCL knockout mice and mature OL numbers are decreased.126 Environmental risk factors for SZ that are related to oxidative stress are also linked to myelination abnormalities. For example, prenatal stress leads to myelination and WM abnormalities,130, 131 and prenatal infection causes effects on myelination and WM.132, 133 The effects of prenatal infection on oxidative stress in adulthood are largely unknown, whereas in young animals the glutathione metabolism is affected.134, 135 Although a link between prenatal infection and both myelination deficits and redox imbalance has thus been observed, it is not clear whether the infection-induced effects on myelination are directly mediated by the redox imbalance. An interesting recent investigation studying the relationship between redox imbalance, reduced myelination and cognition has shown that in vitro hypoxia leads to oxidative stress that causes OPC maturation defects, which can be rescued by free-radical scavengers.136 Likewise, under in vivo hypoxic circumstances ROS levels are higher, OPC maturation does not take place, myelination is decreased, mice show cognitive impairments and when free-radical scavengers are provided the cellular as well as behavioural abnormalities are rescued.136 Hence, redox imbalance causes hypomyelination and cognitive decline.
Redox-related demyelination leads to cognitive defects in MS
The connection between oxidative stress and myelination defects, as observed in SZ, has also been found in MS, a disease associated with major demyelination. For example, in active demyelinating lesions of post mortem MS brains high levels of oxidised lipids and DNA are present, and apoptotic OLs contain oxidised DNA.137, 138 It is thought that in MS the elevated oxidative stress is caused by inflammation and leads to the progressive demyelination that characterises this neurodegenerative disease.139 The fact that MS patients show cognitive symptoms similar to those observed in SZ (for reviews, see Korakaset al.140 and Cardoso et al.141) together with the observation that MS is associated with oxidative stress, decreased myelination and cognitive decline strengthens our hypothesis that an interaction between these factors exists in SZ.
Is hypomyelination during SZ disease onset PFC-specific?
Frontal WM development coincides with the prodromal SZ phase/onset of psychosis
WM maturation commences in central and extends to more lateral brain regions over time,142, 143 and WM volume peaks during early adolescence.144 From this period onwards, the PFC white/grey matter ratio rises with increasing age.145 In frontal areas, WM and connectivity maturation occurs during late adolescence. In addition, the superior longitudinal fasciculus shows increasing connectivity during adolescence146 and corticosubcortical WM tracts reach peaks of maturation between the ages of 23 and 39.147 These findings indicate that WM maturation in frontal areas is ongoing during SZ disease onset.
High-risk individuals have a lower FA than controls,51 and prodromal patients (at-risk individuals who proceed to psychosis) show a progressive reduction in WM integrity in frontal regions over time,51 in contrast to the increase in integrity leading to the WM maturation peak observed in controls.148, 149, 150 WM tracts of other association areas (for example, the uncinate and arcuate fasciculi, the anterior and dorsal cingulate and parts of the corpus callosum) are not different in high-risk versus prodromal individuals.151 Moreover, in prodromal SZ patients WM integrity reductions are observed only in frontal areas, whereas in first-episode patients decreases in WM are found in frontal as well as more caudal regions, including the inferior longitudinal fasciculus and the internal capsule.51, 52, 53 In chronic SZ, lower FA is found in frontal, caudal and more posterior regions, including the corpus callosum, minor and major forceps, inferior fronto-occipital fasciculus and the splenium.50, 54, 55 Thus, even before SZ disease onset a reduced WM integrity occurs in frontal areas that advances in further stages of the disorder, proceeding from frontal towards more caudal and posterior brain regions.
Myelination of most brain regions is completed within the first year of life, whereas the myelination of association areas is ongoing until the thirties, after which myelin levels stabilise and finally decline from the late fifties onwards.152, 153 The extent of cortical myelination is positively correlated with cognitive performance throughout life.153 PFC myelination, which occurs during late adolescence, displays a time frame similar to that of PFC WM development.154 In addition, human PFC myelin-related mRNA expression peaks during late adolescence.155 Thus, the prodromal phase/onset of SZ coincides with the time frame of PFC myelination, and during this stage frontal WM is affected.149 Furthermore, adult SZ dorsolateral and medial PFC mRNA expression patterns of OL-related genes are similar to those in the juvenile healthy developing brain.156 Therefore, it seems that myelin does not reach the mature state in the SZ PFC during adolescence, as it does in healthy brain development.
Cognitive symptomatology in SZ is associated with age-related decline in WM integrity
It is important to note that cognitive symptoms of SZ are observed already during the prodromal phase and worsen when psychosis starts. As such, these symptoms follow a developmental pattern that is similar to the decline in WM integrity in SZ. WM maturation and the cognitive functioning of inhibitory control are indeed correlated.157 In addition, the poor working memory of SZ patients correlates with a low WM integrity in the superior longitudinal fasciculus, a frontal structure that matures during adolescence.157
The role of OPCs in the PFC and other brain areas during adolescence
In the adult brain, OPCs are necessary for myelin repair following damage.158 However, as OPCs make up to 4% of the adult brain159 and appear to be evenly distributed throughout the brain, it seems unlikely that they would be involved in only myelin repair. It has been hypothesised that following the major myelination event during the first year of life a subset of OPCs change into a subtype with a morphology and function different from those of precursor cells of OLs.160, 161 This second type of OPC may have a role in the monitoring of neuronal activity and the immune response.160, 162 Recently, a brain region-dependent variation in the distribution of various subtypes of OPCs has been shown, which differs between young and adult animals.163 For example, adult monkey motor cortex OPCs mainly give rise to perivascular cells, not OLs.164 Likewise, during adolescence, readily myelinated brain areas may well have a set of OPCs that is functionally different from the set of OPCs in brain areas in which myelination is ongoing, such as the PFC that is likely to have OPCs programmed to become OLs.
Oxidative stress may cause apoptosis of pre-OLs in the SZ PFC
The cells that are in transition from OPC to OL are called pre-OLs. The detrimental effects of ROS are the largest in this subtype of OLs.165, 166 The excessive build-up of ROS in pre-OLs during SZ adolescence may lead to apoptosis or a cell cycle arrest followed by an inability to sufficiently produce myelin. In the SZ PFC, a lower number of cells expressing OLIG2 (a marker for all cells of the OL lineage) is observed, with no changes in the number of OPCs, suggesting that indeed PFC OPC maturation impairment in SZ is a likely cause of the lack of myelination in this brain area.167
In addition to the PFC, demyelination and a decreased WM integrity have also been observed in the HIP of SZ brains.168, 169, 170, 171 However, HIP WM defects become apparent during first-episode SZ and are fully evident only during the chronic state of SZ,171, 172, 173 and as such their development follows a different time course than the PFC WM defects that occur already in the prodromal phase. Nevertheless, the neurobiological mechanisms causing OL and myelin defects may be similar in the PFC, HIP and other brain areas of SZ patients. Differentiating OPCs are most vulnerable to oxidative stress; therefore, PFC myelination that is dependent on these cells is harmed during early stages of SZ (as discussed above). Oxidative stress levels increase over time and may reach a level at which mature myelinating OLs are also damaged, and thus regions like the HIP and other brain areas, that depend on mature OLs to maintain proper myelination, will be affected during later stages of SZ. Furthermore, oxidative stress may cause perturbation of de novo myelination in the HIP and other brain areas, a possibility that requires further investigation.
Neurobiological link between hypomyelination and interneuron abnormalities
A significant body of evidence suggests that interneuron abnormalities in both the PFC and HIP have an important role in SZ pathology.174, 175, 176, 177, 178, 179 Interneurons in the PFC mature during adolescence.179 Apart from OLs and OPCs, interneurons are also relatively vulnerable to the effects of oxidative stress because of their high mitochondrial demand.180 Interestingly, oxidative stress-based animal models for SZ display both myelin abnormalities and interneuron defects.181 Oxidative stress in PV interneurons has been proposed as a cause of SZ182 and PV interneuron densities are reduced in, among other brain regions, SZ PFC183 and HIP.184 Impaired myelination of the PV interneurons may render them more susceptible to degeneration in late adolescence, contributing to the reduced PV interneuron densities. Thus, the combination of aberrant myelination and reduction in the number of PV interneurons in the PFC and HIP, both caused by oxidative stress, may well lead to an inefficient neuronal network and eventually to SZ-like symptoms (for review, see Steullet et al.185).
PV interneurons are responsible for the cortical high-frequency gamma-band oscillations that are involved in cognitive functioning and disrupted in SZ.186, 187 The degree of myelination is dependent on neuronal activity,83 and PV cells are the most active of all interneurons and the only interneuron subtype to be myelinated.188 Interestingly, a recent review states that the inefficient myelination of specifically PV interneurons, according to our hypothesis caused by high oxidative stress levels, would generate altered gamma-band oscillations and cognitive deficits in SZ.188
Therapeutic implications
The redox-induced prefrontal OPC-dysfunctioning hypothesis of the cognitive symptoms in SZ may have important implications for novel treatment strategies.
Pharmacological manipulations
On the basis of the molecular map of the relationship between oxidative stress and OPC functioning (Figure 2), new preventive strategies for individuals at high risk for SZ could include antioxidant treatment. In this regard, antioxidant treatment is effective in rodent models,189 and decreases symptom severity in SZ patients.180, 190 Therefore, the use of antioxidants, or compounds that generate an increased production of endogenous antioxidants, may be attractive for SZ therapy.
New potential therapeutic targets include components of the mTOR-P70S6K or ERK1/2 pathway (to be activated) and/or AMPK signalling (to be downregulated) in OPCs, and upregulation of the number of PDGFRα receptors in the cell membrane of OPCs. In this respect, increasing mTOR signalling by inducing the upregulation of brain-derived neurotrophic factor (for example, through 1-amino-1,3-dicarboxycyclopentane) may be considered, and the drugs diosgenin and miconazole could be used to boost ERK1/2 signalling.128, 129, 191 Moreover, drugs that are known to increase myelination by mature OLs and that are tested in the MS field (for example, benztropine192) may prove useful for the treatment of cognitive symptoms in SZ as well.
Cognitive behavioural therapy
Cognitive behavioural therapy specific for cognitive deficits in SZ reduces symptom severity and improves cognitive performance.193, 194, 195, 196 As learning and neuronal activation upregulate myelin levels in cortical regions, and WM integrity in SZ is directly linked to cognitive functioning, beneficial cognitive and other neuronal activity-dependent therapies may be, at least in part, mediated by an experience-dependent increase of PFC myelination.83, 197, 198, 199, 200, 201
Conclusions
Here we propose the redox-induced prefrontal OPC-dysfunctioning hypothesis for the aetiology of cognitive symptoms in SZ (Figure 1). This hypothesis states that in SZ a combination of increased ROS levels caused by genetic and/or environmental factors and a decreased ROS clearance caused by a faulty antioxidant system leads to a build-up of ROS in OPCs. ROS may result in the dysfunctioning of OPCs through a number of cellular pathways, including the ERK1/2 and AMPK signalling cascades that cause an inactivation of the mTOR-P70S6K pathway, and hence negatively influence proliferation and differentiation of this cell type (Figure 2). OPC dysfunctioning occurs in late adolescence, during the critical period of PFC myelination. Therefore, in SZ patients the PFC is hypomyelinated, leading to dysconnectivity and the cognitive symptoms of SZ.
A next step would be the testing of the hypothesis proposed here, in both animal models and SZ patients. For instance, animal models for SZ that show both high oxidative stress levels and PFC hypomyelination (such as the APO-SUS rats, and the rodent prenatal infection and hypoxia models) may be treated with antioxidants from a young age onwards to assess whether lowering oxidative stress can (partially) rescue myelination deficits in the PFC, together with PFC-dependent cognitive functioning, and evaluate the width of the therapeutic window. Furthermore, magnetisation transfer ratio and diffusion magnetic resonance imaging may be used to study PFC myelination over time of individuals at high risk to develop SZ, together with PFC-relevant cognitive assessment. Such studies would establish a relationship between SZ risk, SZ development, PFC WM integrity, myelin levels and cognitive (dys)functioning. In addition, the studies would give insight into whether PFC WM and myelin deficits are indeed caused by a deficiency in prefrontal myelination within the window of SZ disease onset, and whether these shortcomings correlate with cognitive dysfunction in SZ. If confirmed, our hypothesis may significantly contribute to the development of novel antioxidant- and promyelination-based strategies to treat the cognitive symptomatology of this devastating disorder.
References
Orellana G, Slachevsky A . Executive functioning in schizophrenia. Front Psychiatry 2013; 4: 35.
Senkowski D, Gallinat J . Dysfunctional prefrontal gamma-band oscillations reflect working memory and other cognitive deficits in schizophrenia. Biol Psychiatry 2015; 77: 1010–1019.
Zuckerman L, Rehavi M, Nachman R, Weiner I . Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology 2003; 28: 1778–1789.
Brown AS, Patterson PH . Maternal infection and schizophrenia: implications for prevention. Schizophr Bull 2011; 37: 284–290.
Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M . Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav Immun 2012; 26: 623–634.
Paintlia MK, Paintlia AS, Contreras MA, Singh I, Singh AK . Lipopolysaccharide-induced peroxisomal dysfunction exacerbates cerebral white matter injury: attenuation by N-acetyl cysteine. Exp Neurol 2008; 210: 560–576.
Clelland JD, Read LL, Drouet V, Kaon A, Kelly A, Duff KE et al. Vitamin D insufficiency and schizophrenia risk: evaluation of hyperprolinemia as a mediator of association. Schizophr Res 2014; 156: 15–22.
Crews M, Lally J, Gardner-Sood P, Howes O, Bonaccorso S, Smith S et al. Vitamin D deficiency in first episode psychosis: a case-control study. Schizophr Res 2013; 150: 533–537.
McGrath JJ, Eyles DW, Pedersen CB, Anderson C, Ko P, Burne TH et al. Neonatal vitamin D status and risk of schizophrenia: a population-based case-control study. Arch Gen Psychiatry 2010; 67: 889–894.
Eyles D, Almeras L, Benech P, Patatian A, Mackay-Sim A, McGrath J et al. Developmental vitamin D deficiency alters the expression of genes encoding mitochondrial, cytoskeletal and synaptic proteins in the adult rat brain. J Steroid Biochem Mol Biol 2007; 103: 538–545.
Malaspina D, Corcoran C, Kleinhaus KR, Perrin MC, Fennig S, Nahon D et al. Acute maternal stress in pregnancy and schizophrenia in offspring: a cohort prospective study. BMC Psychiatry 2008; 8: 71.
Zhu Z, Li X, Chen W, Zhao Y, Li H, Qing C et al. Prenatal stress causes gender-dependent neuronal loss and oxidative stress in rat hippocampus. J Neurosci Res 2004; 78: 837–844.
Ferreira DJ, da Silva Pedroza AA, Braz GR, da Silva-Filho RC, Lima TA, Fernandes MP et al. Mitochondrial bioenergetics and oxidative status disruption in brainstem of weaned rats: immediate response to maternal protein restriction. Brain Res 2016; 1642: 553–561.
Dalman C, Thomas HV, David AS, Gentz J, Lewis G, Allebeck P . Signs of asphyxia at birth and risk of schizophrenia. Population-based case-control study. Br J Psychiatry 2001; 179: 403–408.
Mirendil H, Thomas EA, De Loera C, Okada K, Inomata Y, Chun J . LPA signaling initiates schizophrenia-like brain and behavioral changes in a mouse model of prenatal brain hemorrhage. Transl Psychiatry 2015; 5: e541.
Zornberg GL, Buka SL, Tsuang MT . Hypoxic-ischemia-related fetal/neonatal complications and risk of schizophrenia and other nonaffective psychoses: a 19-year longitudinal study. Am J Psychiatry 2000; 157: 196–202.
Zugno AI, Pacheco FD, Budni J, de Oliveira MB, Canever L, Heylmann AS et al. Maternal deprivation disrupts mitochondrial energy homeostasis in the brain of rats subjected to ketamine-induced schizophrenia. Metab Brain DIs 2015; 30: 1043–1053.
Akdeniz C, Tost H, Meyer-Lindenberg A . The neurobiology of social environmental risk for schizophrenia: an evolving research field. Soc Psychiatry Psychiatr Epidemiol 2014; 49: 507–517.
Nicodemus KK, Marenco S, Batten AJ, Vakkalanka R, Egan MF, Straub RE et al. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol Psychiatry 2008; 13: 873–877.
Schmidt-Kastner R, van Os J, W M Steinbusch H, Schmitz C . Gene regulation by hypoxia and the neurodevelopmental origin of schizophrenia. Schizophr Res 2006; 84: 253–271.
Chowdari KV, Bamne MN, Nimgaonkar VL . Genetic Association Studies of Antioxidant Pathway Genes and Schizophrenia. Antioxid Redox Signal 2011; 15: 2037–2045.
Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P et al. Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci USA 2007; 104: 16621–16626.
Gravina P, Spoletini I, Masini S, Valentini A, Vanni D, Paladini E et al. Genetic polymorphisms of glutathione S-transferases GSTM1, GSTT1, GSTP1 and GSTA1 as risk factors for schizophrenia. Psychiatr Res 2011; 187: 454–456.
Rodriguez-Santiago B, Brunet A, Sobrino B, Serra-Juhe C, Flores R, Armengol L et al. Association of common copy number variants at the glutathione S-transferase genes and rare novel genomic changes with schizophrenia. Mol Psychiatry 2010; 15: 1023–1033.
Tosic M, Ott J, Barral S, Bovet P, Deppen P, Gheorghita F et al. Schizophrenia and oxidative stress: glutamate cysteine ligase modifier as a susceptibility gene. Am J Hum Genet 2006; 79: 586–592.
Raffa M, Atig F, Mhalla A, Kerkeni A, Mechri A . Decreased glutathione levels and impaired antioxidant enzyme activities in drug-naive first-episode schizophrenic patients. BMC Psychiatry 2011; 11: 124.
Reddy R, Keshavan M, Yao JK . Reduced plasma antioxidants in first-episode patients with schizophrenia. Schizophr Res 2003; 62: 205–212.
Altuntas I, Aksoy H, Coskun I, Caykoylu A, Akcay F . Erythrocyte superoxide dismutase and glutathione peroxidase activities, and malondialdehyde and reduced glutathione levels in schizophrenic patients. Clin Chem Lab Med 2000; 38: 1277–1281.
Li XF, Zheng YL, Xiu MH, Chen da C, Kosten TR, Zhang XY . Reduced plasma total antioxidant status in first-episode drug-naive patients with schizophrenia. Progr Neuropsychopharmacol Biol Psychiatry 2011; 35: 1064–1067.
Do KQ, Trabesinger AH, Kirsten-Kruger M, Lauer CJ, Dydak U, Hell D et al. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci 2000; 12: 3721–3728.
Matsuzawa D, Hashimoto K . Magnetic resonance spectroscopy study of the antioxidant defense system in schizophrenia. Antioxid Redox Signal 2011; 15: 2057–2065.
Gawryluk JW, Wang JF, Andreazza AC, Shao L, Young LT . Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol 2011; 14: 123–130.
Clark D, Dedova I, Cordwell S, Matsumoto I . A proteome analysis of the anterior cingulate cortex gray matter in schizophrenia. Mol Psychiatry 2006; 11: 459–470, 423.
Reyazuddin M, Azmi SA, Islam N, Rizvi A . Oxidative stress and level of antioxidant enzymes in drug-naive schizophrenics. Ind J Psychiatry 2014; 56: 344–349.
Zhang XY, Chen DC, Tan YL, Tan SP, Wang ZR, Yang FD et al. The interplay between BDNF and oxidative stress in chronic schizophrenia. Psychoneuroendocrinology 2015; 51: 201–208.
Sarandol A, Sarandol E, Acikgoz HE, Eker SS, Akkaya C, Dirican M . First-episode psychosis is associated with oxidative stress: Effects of short-term antipsychotic treatment. Psychiatr Clin Neurosci 2015; 69: 699–707.
Miyaoka T, Ieda M, Hashioka S, Wake R, Furuya M, Liaury K et al. Analysis of oxidative stress expressed by urinary level of biopyrrins and 8-hydroxydeoxyguanosine in patients with chronic schizophrenia. Psychiatr Clin Neurosci 2015; 69: 693–698.
Gonzalez-Liencres C, Tas C, Brown EC, Erdin S, Onur E, Cubukcoglu Z et al. Oxidative stress in schizophrenia: a case-control study on the effects on social cognition and neurocognition. BMC Psychiatry 2014; 14: 268.
Dietrich-Muszalska A, Kwiatkowska A . Generation of superoxide anion radicals and platelet glutathione peroxidase activity in patients with schizophrenia. Neuropsychiatr Dis Treat 2014; 10: 703–709.
Ozyurt H, Ozyurt B, Sarsilmaz M, Kus I, Songur A, Akyol O . Potential role of some oxidant/antioxidant status parameters in prefrontal cortex of rat brain in an experimental psychosis model and the protective effects of melatonin. Eur Rev Med Pharmacol Sci 2014; 18: 2137–2144.
Faizi M, Salimi A, Rasoulzadeh M, Naserzadeh P, Pourahmad J . Schizophrenia induces oxidative stress and cytochrome C release in isolated rat brain mitochondria: a possible pathway for induction of apoptosis and neurodegeneration. Iran J Pharm Res 2014; 13 (Suppl): 93–100.
Cruz-Aguado R, Almaguer-Melian W, Diaz CM, Lorigados L, Bergado J . Behavioral and biochemical effects of glutathione depletion in the rat brain. Brain Res Bull 2001; 55: 327–333.
Iguchi Y, Kosugi S, Nishikawa H, Lin Z, Minabe Y, Toda S . Repeated exposure of adult rats to transient oxidative stress induces various long-lasting alterations in cognitive and behavioral functions. PLoS ONE 2014; 9: e114024.
Mamiya T, Kise M, Morikawa K . Ferulic acid attenuated cognitive deficits and increase in carbonyl proteins induced by buthionine-sulfoximine in mice. Neurosci Lett 2008; 430: 115–118.
das Neves Duarte JM, Kulak A, Gholam-Razaee MM, Cuenod M, Gruetter R, Do KQ . N-acetylcysteine normalizes neurochemical changes in the glutathione-deficient schizophrenia mouse model during development. Biol Psychiatry 2012; 71: 1006–1014.
Kulak A, Cuenod M, Do KQ . Behavioral phenotyping of glutathione-deficient mice: relevance to schizophrenia and bipolar disorder. Behav Brain Res 2012; 226: 563–570.
Steullet P, Cabungcal JH, Kulak A, Kraftsik R, Chen Y, Dalton TP et al. Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J Neurosci 2010; 30: 2547–2558.
Wheeler AL, Voineskos AN . A review of structural neuroimaging in schizophrenia: from connectivity to connectomics. Front Hum Neurosci 2014; 8: 653.
Kanaan R, Barker G, Brammer M, Giampietro V, Shergill S, Woolley J et al. White matter microstructure in schizophrenia: effects of disorder, duration and medication. Br J Psychiatry 2009; 194: 236–242.
Liu X, Lai Y, Wang X, Hao C, Chen L, Zhou Z et al. A combined DTI and structural MRI study in medicated-naive chronic schizophrenia. Magn Reson Imaging 2014; 32: 1–8.
Bloemen OJ, de Koning MB, Schmitz N, Nieman DH, Becker HE, de Haan L et al. White-matter markers for psychosis in a prospective ultra-high-risk cohort. Psychol Med 2010; 40: 1297–1304.
Friedman JI, Tang C, Carpenter D, Buchsbaum M, Schmeidler J, Flanagan L et al. Diffusion tensor imaging findings in first-episode and chronic schizophrenia patients. Am J Psychiatry 2008; 165: 1024–1032.
Yao L, Lui S, Liao Y, Du MY, Hu N, Thomas JA et al. White matter deficits in first episode schizophrenia: an activation likelihood estimation meta-analysis. Progr Neuropsychopharmacol Biol Psychiatry 2013; 45: 100–106.
Holleran L, Ahmed M, Anderson-Schmidt H, McFarland J, Emsell L, Leemans A et al. Altered interhemispheric and temporal lobe white matter microstructural organization in severe chronic schizophrenia. Neuropsychopharmacology 2014; 39: 944–954.
Kitamura H, Matsuzawa H, Shioiri T, Someya T, Kwee IL, Nakada T . Diffusion tensor analysis in chronic schizophrenia. A preliminary study on a high-field (3.0 T) system. Eur Arch Psychiatr Clin Neurosci 2005; 255: 313–318.
Nazeri A, Chakravarty MM, Felsky D, Lobaugh NJ, Rajji TK, Mulsant BH et al. Alterations of superficial white matter in schizophrenia and relationship to cognitive performance. Neuropsychopharmacology 2013; 38: 1954–1962.
Muetzel RL, Mous SE, van der Ende J, Blanken LM, van der Lugt A, Jaddoe VW et al. White matter integrity and cognitive performance in school-age children: a population-based neuroimaging study. NeuroImage 2015; 119: 119–128.
Roalf DR, Ruparel K, Verma R, Elliott MA, Gur RE, Gur RC . White matter organization and neurocognitive performance variability in schizophrenia. Schizophr Res 2013; 143: 172–178.
Epstein KA, Cullen KR, Mueller BA, Robinson P, Lee S, Kumra S . White matter abnormalities and cognitive impairment in early-onset schizophrenia-spectrum disorders. J Am Acad Child Adolesc Psychiatry 2014; 53: 362–372, e361-362.
Liu X, Lai Y, Wang X, Hao C, Chen L, Zhou Z et al. Reduced white matter integrity and cognitive deficit in never-medicated chronic schizophrenia: a diffusion tensor study using TBSS. Behav Brain Res 2013; 252: 157–163.
Kuswanto CN, Teh I, Lee TS, Sim K . Diffusion tensor imaging findings of white matter changes in first episode schizophrenia: a systematic review. Clin Psychopharmacol Neurosci 2012; 10: 13–24.
Perez-Iglesias R, Tordesillas-Gutierrez D, McGuire PK, Barker GJ, Roiz-Santianez R, Mata I et al. White matter integrity and cognitive impairment in first-episode psychosis. Am J Psychiatry 2010; 167: 451–458.
Kirkpatrick B, Galderisi S . Deficit schizophrenia: an update. World Psychiatry 2008; 7: 143–147.
Cohen AS, Saperstein AM, Gold JM, Kirkpatrick B, Carpenter WT Jr., Buchanan RW . Neuropsychology of the deficit syndrome: new data and meta-analysis of findings to date. Schizophr Bull 2007; 33: 1201–1212.
Voineskos AN, Foussias G, Lerch J, Felsky D, Remington G, Rajji TK et al. Neuroimaging evidence for the deficit subtype of schizophrenia. JAMA Psychiatry 2013; 70: 472–480.
Spalletta G, De Rossi P, Piras F, Iorio M, Dacquino C, Scanu F et al. Brain white matter microstructure in deficit and non-deficit subtypes of schizophrenia. Psychiatr Res 2015; 231: 252–261.
Rowland LM, Spieker EA, Francis A, Barker PB, Carpenter WT, Buchanan RW . White matter alterations in deficit schizophrenia. Neuropsychopharmacology 2009; 34: 1514–1522.
Galderisi S, Maj M . Deficit schizophrenia: an overview of clinical, biological and treatment aspects. Eur Psychiatry 2009; 24: 493–500.
Karim S, Overshott R, Burns A . Older people with chronic schizophrenia. Aging Mental Health 2005; 9: 315–324.
Loewenstein DA, Czaja SJ, Bowie CR, Harvey PD . Age-associated differences in cognitive performance in older patients with schizophrenia: a comparison with healthy older adults. Am J Geriatr Psychiatry 2012; 20: 29–40.
Assaf Y, Pasternak O . Diffusion tensor imaging (DTI)-based white matter mapping in brain research: a review. J Mol Neurosci 2008; 34: 51–61.
Zhu J, Zhuo C, Qin W, Wang D, Ma X, Zhou Y et al. Performances of diffusion kurtosis imaging and diffusion tensor imaging in detecting white matter abnormality in schizophrenia. NeuroImage 2015; 7: 170–176.
Seal ML, Yucel M, Fornito A, Wood SJ, Harrison BJ, Walterfang M et al. Abnormal white matter microstructure in schizophrenia: a voxelwise analysis of axial and radial diffusivity. Schizophr Res 2008; 101: 106–110.
Scheel M, Prokscha T, Bayerl M, Gallinat J, Montag C . Myelination deficits in schizophrenia: evidence from diffusion tensor imaging. Brain Struct Funct 2013; 218: 151–156.
Du F, Cooper AJ, Thida T, Shinn AK, Cohen BM, Ongur D . Myelin and axon abnormalities in schizophrenia measured with magnetic resonance imaging techniques. Biol Psychiatry 2013; 74: 451–457.
Kubicki M, Park H, Westin CF, Nestor PG, Mulkern RV, Maier SE et al. DTI and MTR abnormalities in schizophrenia: analysis of white matter integrity. NeuroImage 2005; 26: 1109–1118.
Palaniyappan L, Al-Radaideh A, Mougin O, Gowland P, Liddle PF . Combined white matter imaging suggests myelination defects in visual processing regions in schizophrenia. Neuropsychopharmacology 2013; 38: 1808–1815.
Lang DJ, Yip E, MacKay AL, Thornton AE, Vila-Rodriguez F, MacEwan GW et al. 48 echo T(2) myelin imaging of white matter in first-episode schizophrenia: evidence for aberrant myelination. NeuroImage Clin 2014; 6: 408–414.
Baumann N, Pham-Dinh D . Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 2001; 81: 871–910.
Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD . NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 2000; 20: 6404–6412.
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 2008; 11: 1392–1401.
Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A et al. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat Neurosci 2015; 18: 628–630.
Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014; 344: 1252304.
Liu J, Dietz K, DeLoyht JM, Pedre X, Kelkar D, Kaur J et al. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat Neurosci 2012; 15: 1621–1623.
Makinodan M, Rosen KM, Ito S, Corfas G . A critical period for social experience–dependent oligodendrocyte maturation and myelination. Science 2012; 337: 1357–1360.
Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI . Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res 2004; 67: 269–275.
Vostrikov VM, Uranova NA, Rakhmanova VI, Orlovskaia DD . [Lowered oligodendroglial cell density in the prefrontal cortex in schizophrenia]. Zh Nevrol Psikhiatr Im Korsakova 2004; 104: 47–51.
Hof PR, Haroutunian V, Friedrich VL Jr., Byne W, Buitron C, Perl DP et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003; 53: 1075–1085.
Stark AK, Uylings HB, Sanz-Arigita E, Pakkenberg B . Glial cell loss in the anterior cingulate cortex, a subregion of the prefrontal cortex, in subjects with schizophrenia. Am J Psychiatry 2004; 161: 882–888.
Flynn SW, Lang DJ, Mackay AL, Goghari V, Vavasour IM, Whittall KP et al. Abnormalities of myelination in schizophrenia detected in vivo with MRI, and post-mortem with analysis of oligodendrocyte proteins. Mol Psychiatry 2003; 8: 811–820.
Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001; 98: 4746–4751.
Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL et al. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther 2010; 128: 419–432.
Paulson L, Martin P, Persson A, Nilsson CL, Ljung E, Westman-Brinkmalm A et al. Comparative genome- and proteome analysis of cerebral cortex from MK-801-treated rats. J Neurosci Res 2003; 71: 526–533.
Xiu Y, Kong XR, Zhang L, Qiu X, Chao FL, Peng C et al. White matter injuries induced by MK-801 in a mouse model of schizophrenia based on NMDA antagonism. Anat Record 2014; 297: 1498–1507.
Filiou MD, Teplytska L, Otte DM, Zimmer A, Turck CW . Myelination and oxidative stress alterations in the cerebellum of the G72/G30 transgenic schizophrenia mouse model. J Psychiatr Res 2012; 46: 1359–1365.
Haroutunian V, Katsel P, Dracheva S, Davis KL . The human homolog of the QKI gene affected in the severe dysmyelination "quaking" mouse phenotype: downregulated in multiple brain regions in schizophrenia. Am J Psychiatry 2006; 163: 1834–1837.
Rosenbluth J, Bobrowski-Khoury N . Structural bases for central nervous system malfunction in the quaking mouse: dysmyelination in a potential model of schizophrenia. J Neurosci Res 2013; 91: 374–381.
Gregg JR, Herring NR, Naydenov AV, Hanlin RP, Konradi C . Downregulation of oligodendrocyte transcripts is associated with impaired prefrontal cortex function in rats. Schizophr Res 2009; 113: 277–287.
Voineskos AN, Felsky D, Kovacevic N, Tiwari AK, Zai C, Chakravarty MM et al. Oligodendrocyte genes, white matter tract integrity, and cognition in schizophrenia. Cereb Cortex 2013; 23: 2044–2057.
Prata DP, Kanaan RA, Barker GJ, Shergill S, Woolley J, Georgieva L et al. Risk variant of oligodendrocyte lineage transcription factor 2 is associated with reduced white matter integrity. Hum Brain Mapp 2013; 34: 2025–2031.
Che R, Tang W, Zhang J, Wei Z, Zhang Z, Huang K et al. No relationship between 2',3'-cyclic nucleotide 3'-phosphodiesterase and schizophrenia in the Chinese Han population: an expression study and meta-analysis. BMC Med Genet 2009; 10: 31.
Usui H, Takahashi N, Saito S, Ishihara R, Aoyama N, Ikeda M et al. The 2',3'-cyclic nucleotide 3'-phosphodiesterase and oligodendrocyte lineage transcription factor 2 genes do not appear to be associated with schizophrenia in the Japanese population. Schizophr Res 2006; 88: 245–250.
Aleksic B, Ikeda M, Ishihara R, Saito S, Inada T, Iwata N et al. No association between the oligodendrocyte-related gene PLP1 and schizophrenia in the Japanese population. J Hum Genet 2008; 53: 863–866.
Zai G, King N, Wigg K, Couto J, Wong GW, Honer WG et al. Genetic study of the myelin oligodendrocyte glycoprotein (MOG) gene in schizophrenia. Genes Brain Behav 2005; 4: 2–9.
Consortium SWGotPG. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Thorburne SK, Juurlink BH . Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem 1996; 67: 1014–1022.
Juurlink BH . Response of glial cells to ischemia: roles of reactive oxygen species and glutathione. Neurosci Biobehav Rev 1997; 21: 151–166.
Connor JR, Menzies SL . Relationship of iron to oligodendrocytes and myelination. Glia 1996; 17: 83–93.
Li Z, Dong T, Proschel C, Noble M . Chemically diverse toxicants converge on Fyn and c-Cbl to disrupt precursor cell function. PLoS Biol 2007; 5: e35.
Jana M, Pahan K . Down-regulation of myelin gene expression in human oligodendrocytes by nitric oxide: implications for demyelination in multiple sclerosis. J Clin Cell Immunol 2013; 4.
Padhi BK, Pelletier G . Perturbation of myelin basic protein (Mbp) splice variant expression in developing rat cerebellum following perinatal exposure to methylmercury. Toxicol Lett 2012; 213: 374–380.
Kirkpatrick M, Benoit J, Everett W, Gibson J, Rist M, Fredette N . The effects of methylmercury exposure on behavior and biomarkers of oxidative stress in adult mice. Neurotoxicology 2015; 50: 170–178.
Padhi BK, Rosales M, Pelletier G . Perinatal methylmercury exposure perturbs the expression of Plp1 and Cnp splice variants in cerebellum of rat pups. Neurotoxicology 2015; 48: 223–230.
Liu B, Chen X, Wang ZQ, Tong WM . DNA damage and oxidative injury are associated with hypomyelination in the corpus callosum of newborn Nbn(CNS-del) mice. J Neurosci Res 2014; 92: 254–266.
Bonora M, De Marchi E, Patergnani S, Suski JM, Celsi F, Bononi A et al. Tumor necrosis factor-alpha impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ 2014; 21: 1198–1208.
Zou Y, Jiang W, Wang J, Li Z, Zhang J, Bu J et al. Oligodendrocyte precursor cell-intrinsic effect of Rheb1 controls differentiation and mediates mTORC1-dependent myelination in brain. J Neurosci 2014; 34: 15764–15778.
Cao K, Zheng A, Xu J, Li H, Liu J, Peng Y et al. AMPK activation prevents prenatal stress-induced cognitive impairment: Modulation of mitochondrial content and oxidative stress. Free Radic Biol Med 2014; 75: 156–166.
Paintlia AS, Paintlia MK, Singh AK, Singh I . Modulation of Rho-ROCK signaling pathway protects oligodendrocytes against cytokine toxicity via PPAR-α-dependent mechanism. Glia 2013; 61: 1500–1517.
Chen Y, Lu R, Zheng H, Xiao R, Feng J, Wang H et al. The NFKB1 polymorphism (rs4648068) is associated with the cell proliferation and motility in gastric cancer. BMC Gastroenterol 2015; 15: 21.
Glauert HP, Calfee-Mason K, Li Y, Nilakantan V, Twaroski ML, Tharappel J et al. The role of NF-kappaB in PPARalpha-mediated hepatocarcinogenesis. PPAR Res 2008; 2008: 286249.
Shen Q, Li ZQ, Sun Y, Wang T, Wan CL, Li XW et al. The role of pro-inflammatory factors in mediating the effects on the fetus of prenatal undernutrition: implications for schizophrenia. Schizophr Res 2008; 99: 48–55.
Diz-Chaves Y, Astiz M, Bellini MJ, Garcia-Segura LM . Prenatal stress increases the expression of proinflammatory cytokines and exacerbates the inflammatory response to LPS in the hippocampal formation of adult male mice. Brain Behav Immun 2013; 28: 196–206.
Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M . Redox dysregulation, neurodevelopment, and schizophrenia. Cur Opin Neurobiol 2009; 19: 220–230.
Miyake S, Lupher ML Jr., Druker B, Band H . The tyrosine kinase regulator Cbl enhances the ubiquitination and degradation of the platelet-derived growth factor receptor alpha. Proc Natl Acad Sci USA 1998; 95: 7927–7932.
Heldin CH, Ostman A, Ronnstrand L . Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta 1998; 1378: F79–113.
Monin A, Baumann PS, Griffa A, Xin L, Mekle R, Fournier M et al. Glutathione deficit impairs myelin maturation: relevance for white matter integrity in schizophrenia patients. Mol Psychiatry 2014; 20: 827–838.
Carson RP, Kelm ND, West KL, Does MD, Fu C, Weaver G et al. Hypomyelination following deletion of Tsc2 in oligodendrocyte precursors. Ann Clin Transl Neurol 2015; 2: 1041–1054.
Xiao L, Guo D, Hu C, Shen W, Shan L, Li C et al. Diosgenin promotes oligodendrocyte progenitor cell differentiation through estrogen receptor-mediated ERK1/2 activation to accelerate remyelination. Glia 2012; 60: 1037–1052.
Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 2015; 522: 216–220.
Sarkar S, Craig MC, Dell'Acqua F, O'Connor TG, Catani M, Deeley Q et al. Prenatal stress and limbic-prefrontal white matter microstructure in children aged 6-9 years: a preliminary diffusion tensor imaging study. World J Biol Psychiatry 2014; 15: 346–352.
Xu J, Yang B, Yan C, Hu H, Cai S, Liu J et al. Effects of duration and timing of prenatal stress on hippocampal myelination and synaptophysin expression. Brain Res 2013; 1527: 57–66.
Fatemi SH, Folsom TD, Reutiman TJ, Abu-Odeh D, Mori S, Huang H et al. Abnormal expression of myelination genes and alterations in white matter fractional anisotropy following prenatal viral influenza infection at E16 in mice. Schizophr Res 2009; 112: 46–53.
Makinodan M, Tatsumi K, Manabe T, Yamauchi T, Makinodan E, Matsuyoshi H et al. Maternal immune activation in mice delays myelination and axonal development in the hippocampus of the offspring. J Neurosci Res 2008; 86: 2190–2200.
Lante F, Meunier J, Guiramand J, De Jesus Ferreira MC, Cambonie G, Aimar R et al. Late N-acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus 2008; 18: 602–609.
Lante F, Meunier J, Guiramand J, Maurice T, Cavalier M, de Jesus Ferreira MC et al. Neurodevelopmental damage after prenatal infection: role of oxidative stress in the fetal brain. Free Radical Biol Med 2007; 42: 1231–1245.
Miyamoto N, Maki T, Pham LD, Hayakawa K, Seo JH, Mandeville ET et al. Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke 2013; 44: 3516–3521.
Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G et al. Oxidative damage in multiple sclerosis lesions. Brain 2011; 134 (Pt 7): 1914–1924.
Schuh C, Wimmer I, Hametner S, Haider L, Van Dam AM, Liblau RS et al. Oxidative tissue injury in multiple sclerosis is only partly reflected in experimental disease models. Acta Neuropathol 2014; 128: 247–266.
Lassmann H, van Horssen J, Mahad D . Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurology 2012; 8: 647–656.
Korakas N, Tsolaki M . Cognitive impairment in multiple sclerosis: a review of neuropsychological assessments. Cogn Behav Neurol 2016; 29: 55–67.
Cardoso M, Olmo NR, Fragoso YD . Systematic review of cognitive dysfunction in pediatric and juvenile multiple sclerosis. Pediatr Neurol 2015; 53: 287–292.
Cancelliere A, Mangano FT, Air EL, Jones BV, Altaye M, Rajagopal A et al. DTI values in key white matter tracts from infancy through adolescence. AJNR Am J Neuroradiol 2013; 34: 1443–1449.
Yap QJ, Teh I, Fusar-Poli P, Sum MY, Kuswanto C, Sim K . Tracking cerebral white matter changes across the lifespan: insights from diffusion tensor imaging studies. J Neural Transm 2013; 120: 1369–1395.
Pfefferbaum A, Mathalon DH, Sullivan EV, Rawles JM, Zipursky RB, Lim KO . A quantitative magnetic resonance imaging study of changes in brain morphology from infancy to late adulthood. Arch Neurol 1994; 51: 874–887.
Sowell ER, Trauner DA, Gamst A, Jernigan TL . Development of cortical and subcortical brain structures in childhood and adolescence: a structural MRI study. Dev Med Child Neurol 2002; 44: 4–16.
Peters BD, Szeszko PR, Radua J, Ikuta T, Gruner P, DeRosse P et al. White matter development in adolescence: diffusion tensor imaging and meta-analytic results. Schizophr Bull 2012; 38: 1308–1317.
Kochunov P, Williamson DE, Lancaster J, Fox P, Cornell J, Blangero J et al. Fractional anisotropy of water diffusion in cerebral white matter across the lifespan. Neurobiol Aging 2012; 33: 9–20.
Carletti F, Woolley JB, Bhattacharyya S, Perez-Iglesias R, Fusar Poli P, Valmaggia L et al. Alterations in white matter evident before the onset of psychosis. Schizophr Bull 2012; 38: 1170–1179.
Karlsgodt KH, Jacobson SC, Seal M, Fusar-Poli P . The relationship of developmental changes in white matter to the onset of psychosis. Curr Pharm Des 2012; 18: 422–433.
Karlsgodt KH, Niendam TA, Bearden CE, Cannon TD . White matter integrity and prediction of social and role functioning in subjects at ultra-high risk for psychosis. Biol Psychiatry 2009; 66: 562–569.
Peters BD, Dingemans PM, Dekker N, Blaas J, Akkerman E, van Amelsvoort TA et al. White matter connectivity and psychosis in ultra-high-risk subjects: a diffusion tensor fiber tracking study. Psychiatr Res 2010; 181: 44–50.
Siu CR, Balsor JL, Jones DG, Murphy KM . Classic and Golli Myelin Basic Protein have distinct developmental trajectories in human visual cortex. Front Neurosci 2015; 9: 138.
Grydeland H, Walhovd KB, Tamnes CK, Westlye LT, Fjell AM . Intracortical myelin links with performance variability across the human lifespan: results from T1- and T2-weighted MRI myelin mapping and diffusion tensor imaging. J Neurosci 2013; 33: 18618–18630.
Paus T . Mapping brain maturation and cognitive development during adolescence. Trends Cogn Sci 2005; 9: 60–68.
Harris LW, Lockstone HE, Khaitovich P, Weickert CS, Webster MJ, Bahn S . Gene expression in the prefrontal cortex during adolescence: implications for the onset of schizophrenia. BMC Med Genet 2009; 2: 28–28.
Hagihara H, Ohira K, Takao K, Miyakawa T . Transcriptomic evidence for immaturity of the prefrontal cortex in patients with schizophrenia. Mol Brain 2014; 7: 41–41.
Karlsgodt KH, van Erp TG, Poldrack RA, Bearden CE, Nuechterlein KH, Cannon TD . Diffusion tensor imaging of the superior longitudinal fasciculus and working memory in recent-onset schizophrenia. Biol Psychiatry 2008; 63: 512–518.
Kucharova K, Stallcup WB . NG2-proteoglycan-dependent contributions of oligodendrocyte progenitors and myeloid cells to myelin damage and repair. J Neuroinflammation 2015; 12: 161.
Dawson MRL, Polito A, Levine JM, Reynolds R . NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci 2003; 24: 476–488.
Polito A, Reynolds R . NG2-expressing cells as oligodendrocyte progenitors in the normal and demyelinated adult central nervous system. J Anat 2005; 207: 707–716.
Greenwood K, Butt AM . Evidence that perinatal and adult NG2-glia are not conventional oligodendrocyte progenitors and do not depend on axons for their survival. Mol Cell Neurosci 2003; 23: 544–558.
Fernandez-Castaneda A, Gaultier A . Adult oligodendrocyte progenitor cells - Multifaceted regulators of the CNS in health and disease. Brain Behav Immun 2016; 57: 1–7.
Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016; 352: 1326–1329.
Stanton GB, Kohler SJ, Boklweski J, Cameron JL, Greenough WT . Cytogenesis in the adult monkey motor cortex: perivascular NG2 cells are the major adult born cell type. J Comp Neurol 2015; 523: 849–868.
Gerstner B, Buhrer C, Rheinlander C, Polley O, Schuller A, Berns M et al. Maturation-dependent oligodendrocyte apoptosis caused by hyperoxia. J Neurosci Res 2006; 84: 306–315.
Gerstner B, DeSilva TM, Genz K, Armstrong A, Brehmer F, Neve RL et al. Hyperoxia causes maturation-dependent cell death in the developing white matter. J Neurosci 2008; 28: 1236–1245.
Mauney SA, Pietersen CY, Sonntag KC, Woo TU . Differentiation of oligodendrocyte precursors is impaired in the prefrontal cortex in schizophrenia. Schizophr Res 2015; 169: 374–380.
Dracheva S, Davis KL, Chin B, Woo DA, Schmeidler J, Haroutunian V . Myelin-associated mRNA and protein expression deficits in the anterior cingulate cortex and hippocampus in elderly schizophrenia patients. Neurobiol Dis 2006; 21: 531–540.
Kolomeets NS, Uranova NA . [Pathology of oligodendroglia and myelinated fibers of the hippocampus in schizophrenia (an ultrastructural and morphometric study)]. Zh Nevrol Psikhiatr Im SS Korsakova 2008; 108: 52–60.
Hao Y, Yan Q, Liu H, Xu L, Xue Z, Song X et al. Schizophrenia patients and their healthy siblings share disruption of white matter integrity in the left prefrontal cortex and the hippocampus but not the anterior cingulate cortex. Schizophr Res 2009; 114: 128–135.
Thoma RJ, Monnig M, Hanlon FM, Miller GA, Petropoulos H, Mayer AR et al. Hippocampus volume and episodic memory in schizophrenia. J Int Neuropsychol Soc 2009; 15: 182–195.
Lyu H, Hu M, Eyler LT, Jin H, Wang J, Ou J et al. Regional white matter abnormalities in drug-naive, first-episode schizophrenia patients and their healthy unaffected siblings. Aust N Z J Psychiatry 2015; 49: 246–254.
Bernard JA, Orr JM, Mittal VA . Abnormal hippocampal-thalamic white matter tract development and positive symptom course in individuals at ultra-high risk for psychosis. NPJ schizophrenia 2015; 1.
Del Pino I, Garcia-Frigola C, Dehorter N, Brotons-Mas JR, Alvarez-Salvado E, Martinez de Lagran M et al. Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 2013; 79: 1152–1168.
Koh MT, Shao Y, Sherwood A, Smith DR . Impaired hippocampal-dependent memory and reduced parvalbumin-positive interneurons in a ketamine mouse model of schizophrenia. Schizophr Res 2016; 171: 187–194.
Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM et al. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 2013; 68: 184–194.
Schmalbach B, Lepsveridze E, Djogo N, Papashvili G, Kuang F, Leshchyns'ka I et al. Age-dependent loss of parvalbumin-expressing hippocampal interneurons in mice deficient in CHL1, a mental retardation and schizophrenia susceptibility gene. J Neurochem 2015; 135: 830–844.
Stansfield KH, Ruby KN, Soares BD, McGlothan JL, Liu X, Guilarte TR . Early-life lead exposure recapitulates the selective loss of parvalbumin-positive GABAergic interneurons and subcortical dopamine system hyperactivity present in schizophrenia. Transl Psychiatry 2015; 5: e522.
Tseng KY, O'Donnell P . Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex 2007; 17: 1235–1240.
Cabungcal JH, Steullet P, Kraftsik R, Cuenod M, Do KQ . Early-life insults impair parvalbumin interneurons via oxidative stress: reversal by N-acetylcysteine. Biol Psychiatry 2013; 73: 574–582.
Wischhof L, Irrsack E, Osorio C, Koch M . Prenatal LPS-exposure – a neurodevelopmental rat model of schizophrenia – differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Progr Neuropsychopharmacol Biol Psychiatry 2015; 57: 17–30.
Behrens MM, Sejnowski TJ . Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex? Neuropharmacology 2009; 57: 193–200.
Beasley CL, Zhang ZJ, Patten I, Reynolds GP . Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry 2002; 52: 708–715.
Zhang Z, Sun J, Reynolds GP . A selective reduction in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia patients. Chin Med J 2002; 115: 819–823.
Steullet P, Cabungcal JH, Monin A, Dwir D, O'Donnell P, Cuenod M et al. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: a "central hub" in schizophrenia pathophysiology? Schizophr Res 2016; 176: 41–51.
Lewis DA, Curley AA, Glausier JR, Volk DW . Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 2012; 35: 57–67.
Kim T, Thankachan S, McKenna JT, McNally JM, Yang C, Choi JH et al. Cortically projecting basal forebrain parvalbumin neurons regulate cortical gamma band oscillations. Proc Natl Acad Sci USA 2015; 112: 3535–3540.
Stedehouder J, Kushner SA . Myelination of parvalbumin interneurons: a parsimonious locus of pathophysiological convergence in schizophrenia. Mol Psychiatry 2017; 22: 4–12.
Cabungcal JH, Counotte DS, Lewis EM, Tejeda HA, Piantadosi P, Pollock C et al. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 2014; 83: 1073–1084.
Arvindakshan M, Ghate M, Ranjekar PK, Evans DR, Mahadik SP . Supplementation with a combination of omega-3 fatty acids and antioxidants (vitamins E and C) improves the outcome of schizophrenia. Schizophr Res 2003; 62: 195–204.
Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, Dreyfus CF . Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci 2014; 34: 8186–8196.
Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013; 502: 327–332.
Landa Y, Mueser KT, Wyka KE, Shreck E, Jespersen R, Jacobs MA et al. Development of a group and family-based cognitive behavioural therapy program for youth at risk for psychosis. Early Int Psychiatry 2015; 10: 511–521.
Weisbrod M, Aschenbrenner S, Pfuller U, Kaiser S, Roesch-Ely D . [Rehabilitation in persons with schizophrenic spectrum disorders: the impact of cognition and cognitive remediation therapy]. Fortschr Neurol Psychiatr 2014; 82: 128–134.
Morrison AP, Turkington D, Pyle M, Spencer H, Brabban A, Dunn G et al. Cognitive therapy for people with schizophrenia spectrum disorders not taking antipsychotic drugs: a single-blind randomised controlled trial. Lancet 2014; 383: 1395–1403.
Lam KC, Ho CP, Wa JC, Chan SM, Yam KK, Yeung OS et al. Metacognitive training (MCT) for schizophrenia improves cognitive insight: a randomized controlled trial in a Chinese sample with schizophrenia spectrum disorders. Behav Res Ther 2015; 64: 38–42.
Hofstetter S, Tavor I, Tzur Moryosef S, Assaf Y . Short-term learning induces white matter plasticity in the fornix. J Neurosci 2013; 33: 12844–12850.
McKenzie IA, Ohayon D, Li H, Paes de Faria J, Emery B, Tohyama K et al. Motor skill learning requires active central myelination. Science 2014; 346: 318–322.
Zatorre RJ, Fields RD, Johansen-Berg H . Plasticity in gray and white: neuroimaging changes in brain structure during learning. Nat Neurosci 2012; 15: 528–536.
Bennett IJ, Madden DJ . Disconnected aging: cerebral white matter integrity and age-related differences in cognition. Neuroscience 2013; 276: 187–205.
Karbasforoushan H, Duffy B, Blackford JU, Woodward ND . Processing speed impairment in schizophrenia is mediated by white matter integrity. Psychol Med 2015; 45: 109–120.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Maas, D., Vallès, A. & Martens, G. Oxidative stress, prefrontal cortex hypomyelination and cognitive symptoms in schizophrenia. Transl Psychiatry 7, e1171 (2017). https://doi.org/10.1038/tp.2017.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.138
This article is cited by
-
Variations to plasma H2O2 levels and TAC in chronical medicated and treatment-resistant male schizophrenia patients: Correlations with psychopathology
Schizophrenia (2024)
-
Functional myelin in cognition and neurodevelopmental disorders
Cellular and Molecular Life Sciences (2024)
-
Importance of the dysregulation of the kynurenine pathway on cognition in schizophrenia: a systematic review of clinical studies
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Characteristics of the Interaction of Microglia and Oligodendrocytes in the White Matter in Continuous Schizophrenia
Neuroscience and Behavioral Physiology (2023)
-
Caught in vicious circles: a perspective on dynamic feed-forward loops driving oxidative stress in schizophrenia
Molecular Psychiatry (2022)
