
Featured
-
-
Article
| Open Access C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity
C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity
Repeat expansion mutation in C9ORF72 is the most common cause of familial ALS. Here, the authors generate motor neurons from cells of patients with C9ORF72 mutations, and characterize changes in gene expression in these motor neurons compared to genetically corrected lines, which suggest that glutamate receptor subunit GluA1 is dysregulated in this form of ALS.
- Bhuvaneish T. Selvaraj
- , Matthew R. Livesey
- & Siddharthan Chandran
-
Article
| Open Access Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation
Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation
Abnormal cytoplasmic aggregates of FUS are a hallmark of some forms of amyotrophic lateral sclerosis (ALS). Here, using neurons derived from patients with FUS-ALS, the authors demonstrate that impairment of PARP-dependent DNA damage signaling is an event that occurs upstream of neurodegeneration and cytoplasmic aggregate formation in FUS-ALS.
- Marcel Naumann
- , Arun Pal
- & Andreas Hermann
-
Article
| Open Access CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts
CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts
Repeat-associated non-AUG (RAN) translation contributes to the pathogenic mechanism of several microsatellite expansion diseases. Here the authors delineate the different steps involved in recruiting the ribosome to initiate G4C2 RAN translation to produce poly-Glycine Alanine, poly-Glycine Proline, and poly-Glycine Arginine repeats.
- Ricardos Tabet
- , Laure Schaeffer
- & Clotilde Lagier-Tourenne
-
Article
| Open Access Endocytosis regulates TDP-43 toxicity and turnover
Endocytosis regulates TDP-43 toxicity and turnover
Impaired turnover of TDP-43 by impaired autophagy or proteasomal function have been suggested to be the cause of TDP-43 accumulation, a hallmark of ALS. Here the authors demonstrate that endocytosis is also important for regulating TDP-43 turnover and toxicity.
- Guangbo Liu
- , Alyssa N. Coyne
- & J. Ross Buchan
-
Article
| Open Access RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response
RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response
A nucleotide repeat expansion in C9orf72 is a common genetic cause of neurodegenerative disorders. Here, the authors provide insight into the molecular mechanism by which this repeat undergoes Repeat-Associated Non-AUG (RAN) translation, implicating the integrated stress response and eIF2α phosphorylation.
- Katelyn M. Green
- , M. Rebecca Glineburg
- & Peter K. Todd
-
Article
| Open Access A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models
A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models
Astrocytes can have protective or detrimental effects on neurons during injury, but the molecular mechanisms that determine these different states are unresolved. Here the authors identify a pathway via neuronal EphB1 that induces neuroprotective signalling in astrocytes through ephrin-B1 mediated STAT3 activation, which is impaired in models of amyotrophic lateral sclerosis.
- Giulia E. Tyzack
- , Claire E. Hall
- & András Lakatos
-
Article
| Open Access HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients
HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients
Amyotrophic lateral sclerosis (ALS) leads to selective loss of motor neurons. Using motor neurons derived from induced pluripotent stem cells from patients with ALS and FUS mutations, the authors demonstrate that axonal transport deficits that are observed in these cells can be rescued by HDAC6 inhibition.
- Wenting Guo
- , Maximilian Naujock
- & Ludo Van Den Bosch
-
Article
| Open Access Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis
Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a rapidly progressing neurodegenerative disease. Here, Wray and colleagues identify association of the GPX3-TNIP1 locus with ALS using cross-ethnic meta-analyses.
- Beben Benyamin
- , Ji He
- & Dongsheng Fan
-
Article
| Open Access SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits
SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits
The RNA for ALS- and frontotemporal dementia-associated C9ORF72 gene is exported from nucleus via an unknown mechanism. This study shows that reduction of nuclear export adaptor SRSF1 can alleviate neuronal cell death and nuclear export of C9ORF72 inDrosophilaand patient-derived induced motor neurons.
- Guillaume M. Hautbergue
- , Lydia M. Castelli
- & Pamela J. Shaw
-
Article
| Open Access Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation
Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation
TDP-43 aggregation is observed in amyotrophic lateral sclerosis. Here the authors combine X-ray crystallography, nuclear magnetic resonance and electron microscopy studies and show that physiological oligomerization of TDP-43 is mediated through its N-terminal domain, which forms functional and dynamic oligomers antagonizing pathologic aggregation.
- Tariq Afroz
- , Eva-Maria Hock
- & Magdalini Polymenidou
-
Article
| Open Access Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity
Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity
Mutations inCHCHD10 have been recently associated with frontotemporal dementia and amyotrophic lateral sclerosis. Here the authors study the functions of endogenous CHCHD10 in Caenorhabditis elegans, primary neurons, and mouse, and show that it normally protects mitochondria and synaptic integrity, and retains TDP-43 in the nucleus.
- Jung-A. A. Woo
- , Tian Liu
- & David E. Kang
-
Article
| Open Access Genetic correlation between amyotrophic lateral sclerosis and schizophrenia
Genetic correlation between amyotrophic lateral sclerosis and schizophrenia
Relatives of patients with amyotrophic lateral sclerosis have an unexpectedly high incidence of schizophrenia. Here, the authors show a genetic link between the two conditions, suggesting shared neurobiological mechanisms.
- Russell L. McLaughlin
- , Dick Schijven
- & Michael C. O’Donovan
-
Article
| Open Access Monitoring peripheral nerve degeneration in ALS by label-free stimulated Raman scattering imaging
Monitoring peripheral nerve degeneration in ALS by label-free stimulated Raman scattering imaging
Sensitive and label-free imaging methods to visualize nerve degeneration are currently lacking. Here authors show that stimulated Raman scattering (SRS) microscopy can be used to monitor peripheral nerve degeneration in mouse models of amyotrophic lateral sclerosis (ALS) and in postmortem tissue from ALS patients.
- Feng Tian
- , Wenlong Yang
- & Kevin Eggan
-
Article
| Open Access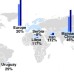 Projected increase in amyotrophic lateral sclerosis from 2015 to 2040
Projected increase in amyotrophic lateral sclerosis from 2015 to 2040
The socioeconomic burden of amyotrophic lateral sclerosis (ALS) is high, but the projected number of cases in the upcoming years is unclear. Here, the authors estimate the number and distribution of ALS cases to 2040, and show that cases are projected to increase, particularly in developing nations.
- Karissa C. Arthur
- , Andrea Calvo
- & Bryan J. Traynor
-
Article
| Open Access Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses
Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses
Abnormal functions of RNA-binding proteins TAF15, FUS and TDP43 are associated with amyotrophic lateral sclerosis. Here, Kapeli et al. characterize the RNA targets of TAF15 and identify points of convergence and divergence between the targets of TAF15, FUS and TDP43 in several neuronal systems.
- Katannya Kapeli
- , Gabriel A. Pratt
- & Gene W. Yeo
-
Article
| Open Access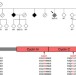 CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia
CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia
Ian Blair and colleagues use genome-wide linkage analysis and whole exome sequencing to identify mutations in the CCNF gene in large cohorts of amyotrophic lateral sclerosis and frontotemporal dementia patients. In addition to validating the mutations in international cohorts, the authors also show that mutant CCNFgene product affects ubiquitination and protein degradation in cultured cells.
- Kelly L. Williams
- , Simon Topp
- & Ian P. Blair
-
Article
| Open Access ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function
ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function
The mechanism by which FUS mutations cause familial ALS remains unclear. Here, the authors use mouse transgenic models to show that a toxic gain-of-function underlies motor neuron degeneration, and that the toxicity of mutant FUS does not depend on a loss or excess of FUS activity.
- Aarti Sharma
- , Alexander K. Lyashchenko
- & Neil A. Shneider
-
Article |
 Neurodegeneration in C. elegans models of ALS requires TIR-1/Sarm1 immune pathway activation in neurons
Neurodegeneration in C. elegans models of ALS requires TIR-1/Sarm1 immune pathway activation in neurons
Abnormal accumulation of TDP-43 and FUS proteins is found in a neurodegenerative disease amyotrophic lateral sclerosis. Here the authors show by modelling the disease in worms that these proteins activate local and distal immune responses, and blocking this pathway in neurons ameliorates the disease.
- Julie Vérièpe
- , Lucresse Fossouo
- & J Alex Parker
-
Article |
 ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP
ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP
Dominant mutations in the RNA-binding protein FUS/TLS cause amyotrophic lateral sclerosis (ALS), an adult-onset motor neuron degenerative disease. Here, the authors show that ALS-causative FUS/TLS mutations directly bind the SMN and U1-snRNP complexes, producing both loss and gain of function effects on RNA processing.
- Shuying Sun
- , Shuo-Chien Ling
- & Don W. Cleveland
-
Article
| Open Access Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability
Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that affects spinal cord motor neurons. Here the authors use induced pluripotent stem cell-derived motor neurons obtained from patients with ALS-linked mutations, and find functional deficits resulting from a progressive decrease in voltage-activated Na+ and K+currents that occur in the absence of cell death.
- Anna-Claire Devlin
- , Karen Burr
- & Gareth B. Miles
-
Article |
 An acetylation switch controls TDP-43 function and aggregation propensity
An acetylation switch controls TDP-43 function and aggregation propensity
The nuclear protein TDP-43 is implicated in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration. Here, Cohen et al. discover lysine acetylation as a modification that regulates TDP-43 function, providing a mechanism that could be implicated in the pathogenesis of TDP-43 proteinopathies.
- Todd J. Cohen
- , Andrew W. Hwang
- & Virginia M. Y. Lee
-
Article |
 An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA–FUS regulatory circuitry
An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA–FUS regulatory circuitry
Abnormal accumulation of the RNA-binding protein FUS and mutations within the FUS gene have been found in association with amyotrophic lateral sclerosis (ALS). Here, Dini Modigliani et al.uncover a FUS regulatory circuit that implicates the microRNAs miR-141 and miR-200a in a feedback loop disrupted by an ALS-associated mutation.
- Stefano Dini Modigliani
- , Mariangela Morlando
- & Irene Bozzoni
-
Article |
 Postnatal muscle modification by myogenic factors modulates neuropathology and survival in an ALS mouse model
Postnatal muscle modification by myogenic factors modulates neuropathology and survival in an ALS mouse model
MyoD and myogenin are muscle regulatory factors that are involved in muscle development. Park et al.study a mouse model of amyotrophic lateral sclerosis and find that MyoD and myogenin have positive and negative effects, respectively, on motor neuron degeneration and muscle denervation.
- Kevin H. J. Park
- , Sonia Franciosi
- & Blair R. Leavitt
-
Article
| Open Access Ligand binding and aggregation of pathogenic SOD1
Ligand binding and aggregation of pathogenic SOD1
Mutations of the SOD1gene are implicated in neurodegenerative diseases such as amyotrophic lateral sclerosis. Wright and colleagues find that SOD1 aggregation in cells is arrested by compounds that bind at the core of SOD1 aggregates, rather than at the dimer interface site.
- Gareth S.A. Wright
- , Svetlana V. Antonyuk
- & S Samar Hasnain
-
Article |
 A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology
A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology
The mislocalization and downregulation of the proteins TDP-43 and ADAR2, respectively, are implicated in amyotrophic lateral sclerosis pathology. Yamashita et al. find that downregulation of ADAR2 results in calcium-permeable AMPA receptor-mediated calpain activation and subsequent aberrant cleavage of TDP-43.
- Takenari Yamashita
- , Takuto Hideyama
- & Shin Kwak
-
Article
| Open Access Structural and molecular insights into the mechanism of action of human angiogenin-ALS variants in neurons
Structural and molecular insights into the mechanism of action of human angiogenin-ALS variants in neurons
Mutations in human angiogenin are implicated in the progression of amyotrophic lateral sclerosis. Thiyagarajan and colleagues show that structural differences between angiogenin variants affect neuronal survival, and the ability to induce stress granules in neuronal cell lines.
- Nethaji Thiyagarajan
- , Ross Ferguson
- & K. Ravi Acharya
 A small-molecule inhibitor of SOD1-Derlin-1 interaction ameliorates pathology in an ALS mouse model
A small-molecule inhibitor of SOD1-Derlin-1 interaction ameliorates pathology in an ALS mouse model