
Abstract
The aim of this study is to analyse cardiac specimens from human cocaine-related overdose, to verify the hypothesis that cardiac toxicity by acute exposure to high dosage of cocaine could be mediated by unbalanced myocardial oxidative stress, and to evaluate the apoptotic response. To address these issues, biochemical and immunohistological markers of oxidative/nitrosative stress were evaluated. We found that i-NOS, NOX2 and nitrotyrosine expression were significantly higher in the hearts of subjects who had died from high doses of cocaine, compared to the control group. Increase of these markers was associated with a dramatic increase in 8-OHdG, another marker of oxidative stress. A high number of TUNEL-positive apoptotic myocells was observed in the study group compared to the control group. The immunoexpression of TNF-α was significantly higher in the cocaine group compared to the control group. Furthermore, we detected a significantly stronger immunoresponse to anti-SMAC/DIABLO in our study group compared to control cases. Both cardiac Fas-dependent and mitochondria-dependent apoptotic pathways appeared to be activated to a greater extent in the cocaine group than in the control group. Our results highlight the central role of oxidative stress in cocaine toxicity. High levels of NOS can promote the oxidation process and lead to apoptosis.
Similar content being viewed by others

Introduction
Cocaine, also known as benzoyl methyl ecgonine, is an alkaloid extracted from erythroxylon coca leaves which is widely abused worldwide1. Acute and chronic cocaine use is responsible for a variety of systemic complications that have been reported in nearly every organ and system: the brain, heart, lungs, kidneys, gastrointestinal tract, musculature, and other organs may be involved2.
In particular, cocaine can cause a kaleidoscope of cardiac pathologies3 and, as its abuse has become widespread, the number of cocaine-related cardiovascular adverse events has dramatically increased4,5,6. The plethora of cocaine-related cardiovascular complications, both acute and chronic, include acute myocardial ischemia and infarction, arrhythmias, sudden death, myocarditis, cardiomyopathy, hypertension, aortic ruptures, and endocarditis7,8,9,10,11.
The cardiac effects of cocaine are complex, and our understanding of the mechanisms of cocaine cardiotoxicity is far from complete. Cocaine cardiotoxicity has long been thought to be mediated indirectly through its sympathomimetic effect, i.e., by inhibiting the presynaptic reuptake and increasing the levels of neuronal catecholamines (dopamine and norepinephrine) with a resulting increase in their concentration in the synaptic cleft and enhanced post-synaptic transmission, as well as enhanced central sympathetic outflow12. The mechanisms of cocaine cardiotoxicity further include blockage of sodium and potassium channels, disruption of excitation-contraction coupling, and altered calcium flux across myocyte cell membrane13.
In recent years, an important area of study has addressed the sources and effect of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in heart diseases, both of which are considered major biologically relevant redox active molecules14. Accumulating evidence suggests that cocaine administration is associated with severe nitrosative/oxidative stress and mitochondrial dysfunction of the heart, and experimental studies have reported altered oxidative balance in the myocardium of chronic cocaine-treated animals15,16,17,18,19,20,21,22,23.
The adrenergic over-stimulation induced by cocaine is correlated to its ability to increase oxidative stress and several mechanisms have been proposed. Previous studies have shown that oxidation of catecholamines results in the formation of highly toxic substances such as aminochromes (e.g. adrenochrome). Adrenochrome is a likely candidate for such a process of redox cycling, leading to the formation of ROS. Acting on different types of heart membranes, ROS cause depletion of cellular antioxidants (e.g. ascorbic acid, AA; glutathione, GSH), intracellular Ca2+ overload, lipid peroxidation and myocardial cell damage24.
Recently it has been hypothesized that oxidative stress could play a significant role in the pathogenesis of cardiotoxicity in chronic cocaine abusers23,25,26. It is noteworthy that ROS, generated during oxidative stress, are responsible for the pathophysiology of various cardiovascular disorders including atherosclerosis, cardiac hypertrophy, cardiomyopathy, heart failure, ventricular remodelling, ischemia/reperfusion injury and myocardial infarction27. Direct cocaine activation of NADPH (nicotinamide adenine dinucleotide phosphate) oxidases (NOX), secondary activation of xantine oxidase, formation of oxidative metabolites, and adrenoceptor hyperstimulation with auto-oxidation of cathecolamines are the hypothesized sources of cocaine-induced ROS in cardiomyocytes26. Even though various studies report the deleterious effects of chronic cocaine assumption on the oxidative balance of the heart, there is a lack of information in the literature about the effects of acute high dosage of cocaine on cardiac oxidative homeostasis.
In the light of these previous findings, this study reports an evaluation of myocardial oxidative damage, analysing cardiac specimens from subjects who have died suddenly from acute cocaine intoxication. This is in order to verify the hypothesis that cardiac toxicity caused in humans by acute exposure to high doses of cocaine could be mediated, at least in part, by unbalanced myocardial oxidative stress. To this end, we selected cases of young people who had died from a high-level dose of cocaine, for which the term overdose is correctly used. To address these issues, biochemical and immunohistological markers of oxidative/nitrosative stress were evaluated.
Results
Toxicological analysis
All the cases are body packers, i.e. international smugglers who insert or ingest into body orifices small packages of drugs for the purposes of the transport and subsequent retrieval of the drug in a foreign country. All the subjects died from cocaine intoxication and the cocaine blood level at the time of death varied as indicated in Table 1.
In four cases, cocaine was associated only with ethyl alcohol. The levels of alcohol found in the blood ranged from trace to 0.8 g/l. In no case was a concentration of other drugs found in the blood or urine at the time of death considered to be responsible for, or have contributed to death.
Biochemical analysis
To determine the degree of oxidative stress in study-group cardiac specimens, the levels of lipid peroxidation and ascorbic acid, and GSH/GSSG ratio were measured and the results are shown in Table 2.
The level of MDA, an indicator of lipid peroxidation, was significantly elevated (p < 0.05) in study group cardiac specimens compared to the control group. Cardiac cytosolic levels of AA were significantly lower (p < 0.05) in the study group compared to controls. Furthermore, the GSH/GSSG ratio was dramatically lower (p < 0.01) in study group cases compared to controls (Fig. 1).

Cardiac cytosolic levels of AA were significantly lower (p < 0.05) and the GSH/GSSG ratio was dramatically lower (p < 0.01) in the study group cases compared to controls. Results were expressed as mean ± SD. The comparison between groups was conducted using the comparison Student’s t test. A value of p < 0.05 was considered statistically significant. *p < 0.05; **p < 0.01 study group vs control group.
The analysis of these parameters highlights the presence of high oxidative stress in examined cardiac samples.
Histology and immunohistochemistry
The histological and immunohistochemical characteristics of the study group compared to the control group are shown in Table 3.
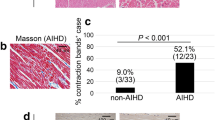
The main change noted during histological observation was the presence of hypereosinophilic, hypercontracted myocardial cells with rhexis of the myofibrillar apparatus into cross-fibre, anomalous, and irregular bands (contraction band necrosis, CBN). This necrosis was, in general, plurifocal and formed by foci found in cardiac specimens of the study group (Fig. 2A). Semi- quantitative analysis showed that the amount of CBN foci was higher in the study group than in the control group.

(A) Hypereosinophilic, hypercontracted myocardial cells with rhexis of the myofibrillar apparatus into cross-fibre, anomalous, and irregular bands (insert: normal aspect of myocytes in control group specimens) (haematoxylin and eosin, original magnification 100x). (B,C) Increase of apoptotic cells was measured by TUNEL assay: values of myocyte cell apoptosis in the cocaine group were significantly higher than control cases. The percentage of apoptotic myocyte nuclei was determined. Apoptosis was measured by the determination of the fraction of myocyte nuclei labelled by TUNEL: 60 ± 13% and 3 ± 1% apoptotic cells were observed in cocaine group and control group (D) (original magnification for B-C-D 63x), respectively. (E) Quantification of apoptosis positive-stained nuclei between groups: the percentage of apoptotic myocyte nuclei was significantly elevated (p < 0.001) in study group specimens compared to the control group. Values are presented as mean ±SD (standard deviation). The unpaired two-way Student’s t-test was used to compare the results obtained for cocaine group with the control group. p < 0.05 was accepted as indicative of significant difference among groups.
Apoptosis was measured by TUNEL assay and values of myocyte cell apoptosis in the study group were significantly higher than control cases. The apoptotic nuclei of myocytes appeared isolated or irregularly distributed over the entire cardiac section from the subepicardium to subendocardium. The percentage of apoptotic myocyte nuclei was determined. The immunohistochemical study revealed an intensive positive result to TUNEL assay (Fig. 2B,C): 60 ± 13% and 3 ± 1% apoptotic cells were observed in cocaine group and control group, respectively.
Semi-quantitative evaluation (grade 0–4) revealed increased myocyte immunoreaction of NOX2 (Fig. 3).

Intense and massive positive (brown in A and in B) myocyte immunoreaction of NOX2 (A,B) in the cocaine group, with representative images of NOX2 immunostaining demonstrated by brown reaction in the cardiac cells compared by the negativity in the control group (C) (original magnification for A-B-C 40x).
i-NOS expression was significantly increased in the hearts of cocaine group with respect to control group subjects whose death was from other causes (Fig. 4).

(A,B) Immunohistochemistry analysis of i-NOS showing positive staining (yellow reaction) in cocaine group heart tissue and compared with the control group (original magnification for A-B-C 40x) (C).
Elevation of the nitrotyrosine markers was documented in the cocaine overdose group with respect to control group (Fig. 5).

Intense nitrotyrosine reaction (darker reaction in cardiac cell) in cocaine group study (A,B), compared with control group (original magnification for A-B-C 40x) (C).
Positive 8-OHdG staining was found in 70% of cardiomyocytes nuclei from study group specimens while they were not detected in control group cardiac specimens (Fig. 6).

8-OHdG positive staining in about 70% of cardiomyocytes nuclei (nuclear brown reaction) from study group specimens (A,B), while they were detected in less than 5% in control group cardiac specimens (C) (original magnification for A-B-C 80x). Values are presented as mean ±SD (standard deviation). The unpaired two-way Student’s t-test was used to compare the results obtained for cocaine group with the control group. p < 0.05 was accepted as indicative of significant difference among groups.
Strong TNF-α and SMAC/DIABLO positivity and weak Bcl-2 immunostaining were detected in cardiac specimens of the study group in respect to the control group (Figs 7 and 8).

SMAC Diablo immunostaining in cardiomyocytes nuclei (nuclear brown reaction) from cocaine group specimens (A,B), while they were detected in less than 5% in control group cardiac specimens (C) (original magnification for A-B-C 100x).

(A,B) Increase of Bcl-2 positive (nuclear brown reaction) immunostaining in the cells’nuclei of the cocaine group; positive cardiomyocytes nuclei were significantly higher than control case group (C) (original magnification for A-B-C 80x).
Discussion
In this study, we evaluated a possible increase in oxidative/nitrosative stress in the cardiac samples of subjects who had died following the assumption of high, lethal doses of cocaine. We found i-NOS, NOX2 and nitrotyrosine expression to have significantly increased in the hearts of these subjects with respect to control group subjects whose death was from other causes. The elevation of these markers was associated with a dramatic increase in another marker of oxidative stress, 8-OHdG, thus supporting the fact that an imbalance in oxidative/nitrosative stress is a key factor in cardiac acute cocaine-induced toxicity.
Our results are in line with previous basic and clinical studies that have highlighted the central role of oxidative/nitrosative stress in cocaine toxicity15,16,17,18,19,20,21,22,23.
Most toxic effects of cocaine on molecular levels are mediated by oxidative stress or mitochondrial dysfunction caused by metabolization of noradrenalin or norcocaine23,28,29. Catecholamines are transformed into ‘aminochromes’, which may undergo redox cycling after entering the mitochondria. This leads to the production of a large quantity of ROS29 that can deplete cellular antioxidants such as AA and GSH. In cardiomyocytes and many mammalian cells, GSH, in a reduced form, is considered the major cytosolic redox buffer. When oxidative stress occurs, the GSH/GSSG ratio can rapidly decrease30. AA is a water-soluble electron transport antioxidant present in the cytosolic and in extracellular fluid and considerable attention has been given to its role in the cardiovascular event31. It is able to scavenge a wide variety of radicals, specifically superoxide anion radicals, peroxyl-derived radicals, and hydroxyl radicals. Previous studies have established that GSH is essential for the physiological function of AA because it is required for the reduction of dehydroascorbic acid and there is metabolic redundancy and overlap of the functions of these antioxidants. In fact, AA can spare GSH and thus protect against the effects of GSH deficiency. Although GSH and AA can perform functions in common, each one is likely to participate in reactions that the other cannot do efficiently32,33. The results of our study show a dramatic decrease in the GSH/GSSG ratio and in AA levels combined with an increase in MDA concentrations in cardiac samples of body-packers. This unequivocally shows that high oxidative stress occurred. In turn, calcium overload and oxidative stress promote mitochondrial permeability transition and cardiomocyte death via activation of both the apoptotic and necrotic pathways28. High levels of NOS can promote the oxidation process and lead to apoptosis34. The pivotal role of mitochondrial oxidative stress in cocaine-induced cardiotoxicity is also confirmed by a study by Vergeade et al. which demonstrates a decrease in cardiotoxicity by MitoQ21, a mitochondrial target antioxidant28 (Fig. 9).

Evaluation of myocardial oxidative damage is the hypothesis that cardiac toxicity caused in humans by acute exposure to high dosage of cocaine could be mediated, at least in part, by unbalanced myocardial oxidative stress.
Conclusively, emerging evidences converge towards the role of oxidative stress imbalance in chronic cocaine toxicity.
This is why it is important to investigate the alterations of nitric oxide (NO) and oxidative stratus in the heart after acute, massive cocaine assumption. NO synthesis is activated by one of the three isoforms of nitric oxide syntase (NOS) that are obligate homodimers catalyzing NADPH-dependent oxidation of L-arginine to NO and L-citrulline: NOS1 (neuronal or n-NOS), NOS2 (inducible or i-NOS), and NOS3 (endothelial or e-NOS)35.
i-NOS produces NO in response to a wide array of stimuli, mostly endotoxin and endogenous pro-inflammatory mediators. Moreover, cocaine enhances NOS levels in different organs and also in plasma36. Methylecgonidine (the principal pyrolysis product of crack cocaine base) has been shown to enhance NO production in cultured neonatal rat cardiomyocytes37. The spectrum of actions of NO is quite complex, and its interactions with oxygen or oxygen-related reactive intermediates (e.g. superoxide) yield numerous ROS and RNS. These account for most of the so-called indirect effects attributed to NO through oxidation, nitrosation, and nitrate reactions referred to as oxidative, nitrosative and nitrative stress, respectively38.
NO influences several aspects of cardiomyocytes functioning. The physiological production of NO in the heart maintains coronary vasodilator tone, and inhibits platelet and neutrophil adhesion, thus performing an active role in cardioprotection39. Excessive NO formation is thought to contribute to contractile dysfunction40,41. The role of i-NOS in cardiac function during the development of left ventricular hypertrophy in mice has been investigated, and recent data demonstrate that NO production via i-NOS plays an important role in modulating cardiac function after moderate aortic banding which mimics long-term hypertension in humans42.
Peroxynitrite (ONOO−), which derives from the reaction between NO and superoxide anions is a potent, citotoxic oxidant capable of inducing all three forms of stress mentioned above. Peroxynitrite-mediated nitrative stress induces severe damage to proteins, lipids, and DNA, resulting in cell damage or apoptosis and cytotoxicity. Nitrotyrosine (NT) formation has been used extensively as a marker of the generation of peroxynitrite43,44,45,46,47,48.
Interestingly, it has been recently shown that oxidative myocardial damage in human cocaine-related cardiomyopathy is mediated by increased NT production26.
Here we present our results showing significantly increased cardiac immunoreactions to NT and i-NOS after massive assumption of cocaine, suggesting an additional mechanism of acute cardiac injury from cocaine.
To obtain further insight into cocaine myocardial damage we explored the expression of NOX2 in cardiomyocytes. Other sources of ROS production are NADPH oxidases (NOX). These enzymes may be responsible for large amounts of superoxide and hydrogen peroxide production in several pathological conditions. The family of NOX proteins plays an integral role in the homeostatic functions of the cell, including gene expression, cell migration, proliferation, senescence and inflammation. The balance between NOX-derived reactive oxygen species production and their elimination by dismutase enzymes is a critical finely-tuned process. The role of NOX2 in human pathologies has been well-documented for specific diseases49, such as diabetes50,51,52,53,54. An increase in the expression of NOX2 in human cardiomyocytes in acute myocardial infarction has been detected55. Finally, chronic binge cocaine consumption has been demonstrated to significantly up-regulate the protein transcription of NOX2 and its component and strongly promote ROS production from NOX2 oxidase20.
Our results showed a significant increase in the immunoexpression of NOX2 in the study group in respect to control cases, thus confirming an imbalance of oxidative status.
Oxidation damage to DNA is the most detrimental, since replication of damaged DNA can lead to genetic mutations or apoptosis56,57. 8-hydroxy-2′-deoxyguanosine (8-OHdG) is a marker of oxidative DNA damage58,59,60,61. Recently the association between 8-OhdG and cardiovascular diseases has been reviewed62, and high levels of this marker in blood and urine have been associated with cardiovascular disease, such as atherosclerosis, heart failure, and ischemic/reperfusion injury following acute myocardial infarction63.
In this study, we assessed the level of DNA oxidative stress by detecting the expression of 8-OHdG. It was found that the positive rate of 8-OHdG expression in myocardial nuclei was higher in the study group than in the control group.
Finally, we explored myocyte apoptosis in our study group compared with control cases.
Apoptosis, a programmed, physiological cellular death, may contribute to many cardiac disorders. Studies have shown that oxidative stress may result in cardiomyopathy through activation of the apoptosis programme in myocardial cells64. Our group’s previous studies indicated that chronic cocaine exposure induces apoptosis in the heart3,22,24. Furthermore, in-utero cocaine exposure induces cardiac myocyte death in experimental animal models65,66,67. Both the intrinsic and extrinsic apoptotic pathways are involved. While previous studies suggest that cocaine-induced apoptosis in cardiomyocytes is mediated only by the mitochondria-dependent, intrinsic, pathway6,9,68, Liou et al.69 recently demonstrated that chronic cocaine exposure appeared to also activate the cardiac Fas- dependent, extrinsic apoptosis.
To investigate the two apoptotic pathways in our study group, TNF-α (an upstream component of cardiac Fas-dependent apoptotic signalling pathway), Bcl-2 and SMAC/Diablo (as markers of mitochondria-dependent apoptotic pathway) were examined.
The immunoexpression of TNF-α was significantly higher in the cocaine group compared to the control group, thus suggesting that also in acute cardiac toxicity from cocaine, the Fas-dependent pathway could be a key factor in triggering cardiac apoptosis.
Furthermore, we detected a significantly stronger immunoresponse to antibody anti-SMAC/DIABLO in our study group compared to control cases, thus suggesting the possibility that mitochondrial-induced apoptosis could be triggered in acute cardiotoxicity from cocaine.
SMAC/DIABLO is a mitochondria-derived pro-apoptotic molecule that appears to function by neutralizing the caspase-inhibitory properties of the IAP family of proteins, particularly XIAP70,71. Similarly to cytochrome c, SMAC/DIABLO is encoded by a nuclear gene and is subsequently imported into mitochondria. Mitochondrial SMAC/DIABLO release is a general feature of apoptosis, and Bcl-2 regulates this event. In cells stimulated to die in response to diverse pro-apoptotic agents, including death receptor ligation, cytotoxic drugs and DNA-damaging agents, SMAC/DIABLO accumulation within the cytosol was readily detected. This coincided with a concomitant loss of SMAC/DIABLO from mitochondria. It was demonstrated that Bcl-2 blocks apoptosis-associated release of SMAC/DIABLO from mitochondria, confirming the anti-apoptotic roles of Bcl-272.
Conclusions
Conclusively, our main findings can be summarized as follows: 1) a great number of TUNEL-positive apoptotic myocells was observed in the study group compared to the control group; 2) both cardiac Fas-dependent and mitochondria-dependent apoptotic pathways appeared to be more activated in the cocaine group compared to the control group; 3) myocardial oxidative damage, the evidence for which is based on increases in i-NOS, NOX2, NT, and 8-OHdG expression and an increase in the biochemical marker of lipid peroxidation (MDA) and in the depletion of antioxidant reserve as expressed by AA levels and GSH/GSSG ratio.
Material and Methods
Study population
The processing of the data reported in this paper is covered by the general authorization to process personal data for scientific research purposes granted by the Italian Data Protection Authority (1 March 2012 as published in Italy’s Official Journal no. 72 dated 26 March 2012) since the data do not entail any significant personalized impact on data subjects. Our study does not involve the application of experimental protocols; therefore it does not require approval by an institutional and/or licensing committee. In all cases, local prosecutors opened an investigation, ordering that an autopsy be performed to clarify the exact cause of death. From the autopsies performed at our departments between January 2001 and December 2014, nine cases (six males, three females, mean age 28.1 ± 10,9 years) of acute cocaine intoxication (body-packers and body-stuffers) were selected. These nine subjects comprise our study population (study group). Cases with toxicological findings suggestive of polysubstances abuse were excluded. Results were compared with cardiac samples from nine subjects matched for age and sex, who had died suddenly from traumatic causes (control group) in which toxicological analyses were negative.
Toxicological analysis
Toxicological analyses by solid-liquid extraction and gas chromatography-mass spectrometry (GC-MS) were carried out to identify and quantify any substance present in biological fluid and organs.
Biochemical analysis
Malondialdehyde (MDA) assessment
The extent of lipid peroxidation, a marker of oxidative stress, was estimated using MDA level calculation. Cardiac samples were homogenized in a 0.04 M K+ phosphate buffer (pH 7.4) containing 0.01% butyl hydroxytoluene (BHT) (1:5 w/v, 0 °C) to prevent the artificial oxidation of polyunsaturated free fatty acids during the assay. This homogenate was deproteinised with acetonitrile (1:1) and then centrifuged at 3000 g for 15 min. The supernatants were used for MDA-analysis after pre-column derivatization with 2.4-dinitrophenylhydrazine. The MDA-hydrazone was quantified by isocratic reversed-phase HPLC method with UV detection as described by Shara et al.73.
Reduced glutathione (GSH) and oxidized glutathione (GSSG) determination
Reduced glutathione (GSH) is considered to be one of the most important scavengers of ROS, and its ratio to oxidised glutathione (GSSG) may be used as a marker of oxidative stress. Cardiac tissue was homogenized in ethylenediaminetetraacetic acid (EDTA) K+ phosphate buffer, pH 7.4 (1:3, w/v) at 0 °C and 1 ml aliquots of the samples were added to an equal volume of 25% tricholoroacetic acid (TCA). After centrifugation at 2000 g for 15 min (0 °C), the supernatant was washed with diethyl ether. The levels of total GSH were measured using an enzyme-recycling assay based on the colorimetric reaction of GSH with DTNB in the presence of excess glutathione reductase and NADPH. TNB chromophore formation was followed spectrophotometrically at 405 nm. Total GSH was analyzed as described by Tietze73 and GSSG was determined according to Griffith’s method74.
Ascorbic Acid (AA) assay
Cardiac tissues were homogenized in EDTA-K+ phosphate buffer pH 7.4 (1:4 w/v) at 0 °C and analysed as described by Ross75. Samples (0.6 ml) were added to an equal volume of 10% (w/v) metaphosphoric acid and immediately centrifuged at 2000 × g at 0 °C for 10 min. Ascorbic acid was determined with a simple method by reversed-phase HPLC using an ion-pairing reagent with UV detection at 262 nm.
Histological and immunohistochemical study
In each case, tissue samples were obtained from the heart (seven standard specimens). Paraffin- embedded heart specimens were sectioned at 4 μm and stained with haematoxilin, eosin and Masson trichrome. In addition, an immunohistochemical study was performed with antibodies anti-NT (nitrotyrosine, Santa Cruz, CA, USA), anti-NOX2 (nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2, Santa Cruz, CA, USA), anti-i-NOS (inducible Nitric oxide synthase, Santa Cruz, CA, USA), anti-8-OHdG (8-Hydroxy-2′-deoxyguanosine, JaICA, Japan), anti-TNF-α (tumour necrosis factor-α, Santa Cruz, CA, USA), anti-BCL2 (B-cell lymphoma 2, Chemicon, Millipore, Billerica, MA, USA), anti-SMAC/DIABLO (second mithocondria-derived activator of caspase/direct IAP binding protein with low PI, Chemicon, Millipore, Billerica, MA, USA), and apoptosis with TUNEL assay (Apotag Plus Peroxidase In Situ Apoptosis Detection Kit, Chemicon, Millipore, Billerica, MA, USA).
Briefly, we used 4 μm-thick paraffin sections mounted on slides covered with 3, aminopropyltriethoxysilane (Fluka, Buchs, Switzerland). Pre-treatment was necessary to facilitate antigen retrieval and to increase membrane permeability to antibodies anti-TNF- α, anti-NOX2, anti- 8-OHdG, anti-i-NOS, anti-Nitrotyrosine, anti-BCL2 and anti-SMAC/DIABLO boiling in 0.1 M Citric Acid buffer. The primary antibody was applied in 1:10 ratio for 8-OHdG, in 1:50 ratio for NOX2, i-NOS, BCL2, in 1:100 ratio for Nitrotyrosine and SMAC/DIABLO, in 1:600 ratio for TNF-α, and incubated for 120 min at 20 °C. The detection system utilized was the LSAB + kit (Dako, Copenhagen, Denmark), a refined avidin-biotin technique in which a biotinylated secondary antibody reacts with several peroxidase-conjugated streptavidin molecules. For TUNEL assay (Apotag Plus Peroxidase In Situ Apoptosis Detection Kit, Chemicon (Millipore), Billerica, MA, USA) sections were pre-treated with Proteinase K (Sigma-Aldrich, Buchs, Switzerland) (20 μg/ml) for 15 min. at 20 °C; covered and incubated with the TdT enzyme; diluted in a ratio of 30% in reaction buffer for 60 min. at 38 °C; put in a coplin jar containing working strength stop/wash buffer; shaken for 15 seconds and incubated for 10 minutes at 20 °C, and covered and incubated with anti-digoxigenin conjugate for 30 minutes at 20 °C. The sections were counterstained with Mayer’s haematoxylin, dehydrated, cover slipped and observed in a Leica DM6000 optical microscope (Leica, Cambridge, UK). Apoptosis was measured by TUNEL assay. In order to determine the fraction of myocyte nuclei labelled by TUNEL, the number of myocyte nuclei per unit area of tissue was calculated by counting an average of 10 fields, 1.4 mm2 each, at a magnification of 10x in each area of myocardium sampled. The percentage of apoptotic myocyte nuclei was determined. For semiquantitative analysis, slides were scored in a blind manner by two observers (MN, ET). The intensity of immunopositive expression was assessed semiquantitatively on a scale of 0–4 as follows: 0 = no immunoreactivity, 1 = mild immunopositivity in scattered cells, 2 = immunopositivity in up to one third of cells, 3 = immunopositivity in up to half the cells and 4 = strong immunopositivity in the majority or all of the cells. In cases of divergent scoring, a third observer (VF) decided the final category.
Statistical analysis
Results were expressed as mean ± SD. The comparison between groups was conducted using the comparison Student’s T test. A value of P < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Turillazzi, E. et al. Myocardial oxidative damage is induced by cardiac Fas-dependent and mitochondria-dependent apoptotic pathways, in human cocaine-related overdose. Sci. Rep. 7, 44262; doi: 10.1038/srep44262 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
United Nation Office on Drugs and Crime, World Drug Report 2015. Available at: https://www.unodc.org/documents/wdr2015/World_Drug_Report_2015.pdf. (Accessed: 22th March 2016) (2015).
Riezzo, I. et al. Side effects of cocaine abuse: multiorgan toxicity and pathological consequences. Curr. Med. Chem. 19, 5624–5646 (2012).
Turillazzi, E. et al. Cardiovascular effects of cocaine: cellular, ionic and molecular mechanisms. Curr. Med. Chem. 19, 5664–5676 (2012).
Kloner, R. A. & Rezkalla, S. H. Cocaine and the heart. N. Engl. J. Med. 348, 487–488 (2003).
Lange, R. A., Cigarroa, J. E. & Hillis, L. D. Theodore E. Woodward award: cardiovascular complications of cocaine abuse. Trans. Am. Clin. Climatol. Assoc. 115, 99–111 (2004).
Schwartz, B. G., Rezkalla, S. & Kloner, R. A. Cardiovascular effects of cocaine. Circulation 122, 2558–2569 (2010).
Kloner, R. A., Hale, S., Alker, K. & Rezkalla, S. The effects of acute and chronic cocaine use on the heart. Circulation 85, 407–419 (1992).
Pozner, C. N., Levine, M. & Zane, R. The cardiovascular effects of cocaine. J. Emerg. Med. 29, 173–178 (2005).
Phillips, K. et al. Cocaine cardiotoxicity: a review of the pathophysiology, pathology, and treatment options. Am. J. Cardiovasc. Drugs 9, 177–196 (2009).
Maraj, S., Figueredo, V. M. & Lynn Morris, D. Cocaine and the heart. Clin. Cardiol. 33, 264–269 (2010).
Bhargava, S. & Arora, R. R. Cocaine and cardiovascular complications. Am. J. Ther. 18, e95–e100 (2011).
Afonso, L., Mohammad, T. & Thatai, D. Crack whips the heart: a review of the cardiovascular toxicity of cocaine. Am. J. Cardiol. 100, 1040–1043 (2007).
Stankowski, R. V., Kloner, R. A. & Rezkalla, S. H. Cardiovascular consequences of cocaine use. Trends Cardiovasc. Med. 25, 517–526 (2015).
Costa, V. M. et al. Contribution of catecholamine reactive intermediates and oxidative stress to the pathologic features of heart diseases. Curr. Med. Chem. 18, 2272–2314 (2011).
Moritz, F. et al. Role of reactive oxygen species in cocaine-induced cardiac dysfunction. Cardiovasc. Res. 59, 834–843 (2003).
Pacifici, R. et al. Immunosuppression and oxidative stress induced by acute and chronic exposure to cocaina in rat. Int. Immunopharmacol. 3, 581–592 (2003).
Kovacic, P. Role of oxidative metabolites of cocaine in toxicity and addiction: oxidative stress and electron transfer. Med. Hypotheses 64, 350–356 (2005).
Ren, S. et al. Effect of long-term cocaine use on regional left ventricular function as determined by magnetic resonance imaging. Am. J. Cardiol. 97, 1085–1088 (2006).
Isabelle, M. et al. NADPH oxidase inhibition prevents cocaine-induced up-regulation of xanthine oxidoreductase and cardiacdysfunction. J. Mol. Cell. Cardiol. 42, 326–332 (2007).
Fan, L. et al. Chronic cocaine-induced cardiac oxidative stress and mitogen-activated protein kinase activation: the role of Nox2 oxidase. J. Pharmacol. Exp. Ther. 328, 99–106 (2009).
Vergeade, A. et al. Mitochondrial impairment contributes to cocaine-induced cardiac dysfunction: Prevention by the targeted antioxidant MitoQ. Free Radic. Biol. Med. 49, 748–756 (2010).
Cerretani, D. et al. Role of oxidative stress in cocaine-induced cardiotoxicity and cocaine-related death. Curr. Med. Chem. 19, 5619–5623 (2012).
Liaudet, L., Calderari, B. & Pacher, P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail. Rev. 19, 815–824 (2014).
Fineschi, V. et al. Markers of cardiac oxidative stress and altered morphology after intraperitoneal cocaine injection in a rat model. Int. J. Legal Med. 114, 323–330 (2001).
Costa, V. M., Carvalho, F., Duarte, J. A., Bastos, Mde L. & Remião, F. The heart as a target for xenobiotic toxicity: the cardiac susceptibility to oxidative stress. Chem. Res. Toxicol. 26, 1285–1311 (2013).
Frustaci, A. et al. Oxidative myocardial damage in human cocaine-related cardiomyopathy. Eur. J. Heart Fail. 17, 283–290 (2015).
Jain, A. K., Mehra, N. K. & Swarnakar, N. K. Role of antioxidants for the treatment of cardiovascular diseases: challenges and opportunities. Curr. Pharm. Des. 21, 4441–4455 (2015).
Finsterer, J. & Ohnsorge, P. Influence of mitochondrion-toxic agents on the cardiovascular system. Regul. Toxicol. Pharmacol. 67, 434–445 (2013).
Varga, Z. V., Ferdinandy, P., Liaudet, L. & Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 309, H1453–1467 (2015).
Palace, V., Kumar, D., Hill, M. F., Khaper, N. & Singal, P. K. Regional differences in non-enzymatic antioxidants in the heart under control and oxidative stress conditions. J. Mol. Cell. Cardiol. 31, 193–202 (1999).
Ferrari, R. et al. Oxygen free radicals and myocardial damage: protective role of thiol-containing agents. Am. J. Med. 91, 95S–105S (1991).
Mårtensson, J. & Meister, A. Gluthatione deficiency decreases tissue ascorbate levels in newborn rats: ascorbate spares glutatione and protects. Proc. Natl. Acad. Sci. USA 88, 4656–4660 (1991).
Mårtensson, J., Han, J., Griffith, O. W. & Meister, A. Glutathione ester delays the onset of scurvy in ascorbate-deficient guinea pigs. Proc. Natl. Acad. Sci. USA 90, 317–321 (1993).
Bagetta, G. et al. Inducible nitric oxide synthase is involved in the mechanisms of cocaine enhanced neuronal apoptosis induced by HIV-1 gp120 in the neocortex of rat. Neurosci. Lett. 356, 183–186 (2004).
Förstermann, U. & Sessa, W. C. Nitric oxide synthases: regulation and function. Eur. Heart J. 33, 829–837 (2012).
Elliott, J. C., Ijames, S. G. & Lysle, D. T. Cocaine increases inducible nitric oxide synthase expression in rats: effects of acute and binge administration. Int. Immunopharmacol. 3, 1011–1018 (2003).
Yang, Y. et al. Enhancement of nitric oxide production by methylecgonidine in cultured neonatal rat cardiomyocytes. Br. J. Pharmacol. 135, 188–196 (2002).
Lechner, M., Lirk, P. & Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin. Cancer Biol. 15, 277–289 (2005).
Jones, S. P. & Bolli, R. The ubiquitous role of nitric oxide in cardioprotection. J. Mol. Cell. Cardiol. 40, 16–23 (2006).
Heusch, P. et al. Increased inducible nitric oxide synthase and arginase II expression in heart failure: no net nitrite/nitrate production and protein S-nitrosylation. Am. J. Physiol. Heart Circ. Physiol. 299, H446–453 (2010).
Soskić, S. S. et al. regulation of inducible nitric oxide synthase (iNOS) and its potential role in insulin resistance, diabetes and heart failure. Open Cardiovasc. Med. J. 5, 153–163 (2011).
Dias, F. A. et al. Ablation of iNOS delays cardiac contractile dysfunction in chronic hypertension. Front. Biosci. (Elite Ed.) 2, 312–324 (2010).
Beckam, J. S. Oxidative damage and tyrosine nitration from peroxynitrite. Chem. Res. Toxicol. 9, 836–844 (1996).
Szabó, C. Multiple pathways of peroxinitrite citotoxicity. Toxicol. Lett. 140-141, 105–112 (2003).
Xie, Y. W. & Wolin, M. S. Role of nitric oxide and its interaction with superoxide in the suppression of cardiac muscle mitochondrial respiration: involvement in response to hypoxia/reoxygenation. Circulation 94, 2580–2586 (1996).
Li, Y., Sarkar, O., Brochu, M. & Anand-Srivastava, M. B. Natriuretic peptide receptor-C attenuates hypertension in spontaneously hypertensive rats: role of nitroxidative stress and Gi proteins. Hypertension 63, 846–855 (2014).
Uppu, R. M. et al. Cardiovascular effects of peroxynitrite. Clin Exp Pharmacol Physiol. 34, 933–937 (2007).
Pacher, P., Beckman, J. S. & Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424 (2007).
Gray, S. P. & Jandeleit-Dahm, K. A. The role of NADPH oxidase in vascular disease - hypertension, atherosclerosis & stroke. Curr. Pharm. Des. 21, 5933–5944 (2015).
Görbe, A. et al. Cholesterol diet leads to attenuation of ischemic preconditioning-induced cardiac protection: the role of connexin 43. Am. J. Physiol. Heart Circ. Physiol. 300, H1907–H1913 (2011).
Guo, J. et al. Cardioprotection against doxorubicin by metallothionein Is associated with preservation of mitochondrial biogenesis involving PGC-1α pathway. Eur. J. Pharmacol. 737, 117–124 (2014).
Hu, C. et al. Chronic ethanol consumption increases cardiomyocyte fatty acid uptake and decreases ventricular contractile function in C57BL/6J mice. J. Mol. Cell Cardiol. 59, 30–40 (2013).
Ichikawa, Y. et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 124, 617–630 (2014).
Gardner, J. D. & Mouton, A. J. Alcohol effects on cardiac function. Compr. Physiol. 5, 791–802 (2015).
Krijnen, P. A. et al. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol. 56, 194–199 (2003).
Evans, M. D., Dizdaroglu, M. & Cooke, M. S. Oxidative DNA damage and diseases: induction, repair and significance. Mutat. Res. 567, 1–61 (2004).
Cooke, M. S. & Evans, M. D. 8-Oxo-deoxyguanosine: reduce, reuse, recycle? Proc. Natl. Acad. Sci. USA 104, 13535–13536 (2007).
Fraga, C. G., Shigenaga, M. K., Park, J. W., Degan, P. & Ames, B. N. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 87, 4533–4537 (1990).
Culter, R. G. Human longevity and aging: possible role of reactive oxygen species. Ann. N. Y. Acad. Sci. 621, 1–28 (1991).
Kasai, H. Chemistry-based studies on oxidative DNA damage: formation, repair, and mutagenesis. Free Radic. Biol. Med. 33, 450–456 (2002).
Valavanidis, A., Vlachogianni, T. & Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health. C Environ. Carcinog. Ecotoxicol. Rev. 27, 120–139 (2009).
Kroese, L. J. & Scheffer, P. G. 8-hydroxy-2′-deoxyguanosine and cardiovascular disease: a systematic review. Curr. Atheroscler. Rep. 16, 452 (2014).
Nagayoshi, Y. et al. Urinary 8-hydroxy-2′-deoxyguanosine levels increase after reperfusion in acute myocardial infarction and may predict subsequent cardiac events. Am. J. Cardiol. 95, 514–517 (2005).
Cesselli, D. et al. Oxidative stress mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 89, 279–286 (2001).
Bae, S. & Zhang, L. Prenatal cocaine exposure increases apoptosis of neonatal rat heart and heart susceptibility to ischemia-reperfusion injury in 1-month-old rat. Br. J. Pharmacol. 144, 900–907 (2005).
Feng, Q. Postnatal consequences of prenatal cocaine exposure and myocardial apoptosis: does cocaine in utero imperil the adult heart? Br. J. Pharmacol. 144, 887–888 (2005).
Sinha-Hikim, I. et al. Minocycline suppresses oxidative stress and attenuates fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure. Apoptosis 16, 563–573 (2011).
Zhang, L., Xiao, Y. & He, J. Cocaine and apoptosis in myocardial cells. Anat. Rec. 257, 208–216 (1999).
Liou, C. M., Tsai, S. C., Kuo, C. H., Ting, H. & Lee, S. D. Cardiac Fas-dependent and mitochondria-dependent apoptosis after chronic cocaine abuse. Int. J. Mol. Sci. 15, 5988–6001 (2014).
Du, C., Fang, M., Li, Y., Li, L. & Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42 (2000).
Verhagen, A. M. et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43–53 (2000).
Adrain, C., Creagh, E. M. & Martin, S. J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 20, 6627–6636 (2001).
Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 27, 502–522 (1969).
Shara, M. A., Dickson, P. H., Bagchi, D. & Stohs, S. J. Excretion of formaldehyde, malondialdehyde, acetaldehyde and acetone in the urine of rats in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin, paraquat, endrin and carbon tetrachloride. J. Chromatogr. 576, 221–233 (1992).
Griffith, O. W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 27, 922–935 (1999).
Ross, M. A. Determination of ascorbic acid and uric acid in plasma by high-performance liquid chromatography. J. Chromatogr. B Biomed. Appl. 657, 197–200 (1994).
Author information
Authors and Affiliations
Contributions
E.T. and V.F. conceived the experiments, D.C., A.I.F., S.C., L.M., M.N., E.P. A.S. and A.V. conducted the experiments, L.C. and M.D.P. and P.F. analysed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Turillazzi, E., Cerretani, D., Cantatore, S. et al. Myocardial oxidative damage is induced by cardiac Fas-dependent and mitochondria-dependent apoptotic pathways in human cocaine-related overdose. Sci Rep 7, 44262 (2017). https://doi.org/10.1038/srep44262
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep44262
This article is cited by
-
Lethal nitrous oxide (N2O) intoxication during surgery: the contribution of immunohistochemistry in identifying the cause of death: a case report
Journal of Medical Case Reports (2023)
-
Behavioral, histopathological, genetic, and organism-wide responses to phenanthrene-induced oxidative stress in Eisenia fetida earthworms in natural soil microcosms
Environmental Science and Pollution Research (2022)
-
Chronic exposure to tramadol induces cardiac inflammation and endothelial dysfunction in mice
Scientific Reports (2021)
-
Tumor- and mitochondria-targeted nanoparticles eradicate drug resistant lung cancer through mitochondrial pathway of apoptosis
Journal of Nanobiotechnology (2020)
-
Changes in gene expression patterns in postmortem human myocardial infarction
International Journal of Legal Medicine (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
