
Abstract
Potassium (K+) deficiency as a common abiotic stress can inhibit the growth of plants and thus reduce the agricultural yields. Nevertheless, scarcely any development has been promoted in wheat transcriptional changes under K+ deficiency. Here we investigated root transcriptional changes in two wheat genotypes, namely, low-K+ tolerant “Tongzhou916” and low-K+ susceptible “Shiluan02-1”. There were totally 2713 and 2485 probe sets displayed expression changes more than 1.5-fold in Tongzhou916 and Shiluan02-1, respectively. Low-K+ responsive genes mainly belonged to the categories as follows: metabolic process, cation binding, transferase activity, ion transporters and so forth. We made a comparison of gene expression differences between the two wheat genotypes. There were 1321 and 1177 up-regulated genes in Tongzhou916 and Shiluan02-1, respectively. This result indicated that more genes took part in acclimating to low-K+ stress in Tongzhou916. In addition, there were more genes associated with jasmonic acid, defense response and potassium transporter up-regulated in Tongzhou916. Moreover, totally 19 genes encoding vacuolar H+-pyrophosphatase, ethylene-related, auxin response, anatomical structure development and nutrient reservoir were uniquely up-regulated in Tongzhou916. For their important role in root architecture, K+ uptake and nutrient storage, unique genes above may make a great contribution to the strong low-K+ tolerance in Tongzhou916.
Similar content being viewed by others
Introduction
Potassium (K+) as the most important macronutrients in the field, takes up about 2 to 10 percent of the total dry weight in plants1. As is known to all, K+ contributes a lot to enzyme activation, osmoregulation, protein synthesis, electrical neutralization, photosynthesis and control of turgor pressure2. Thus K+ deficiency brings about many negative impacts on plants, such as: growth inhibition on account of inappropriate osmotic pressure; nutrition imbalance due to the arrest of photosynthesis and protein synthesis; decrease in pathogen resistance and so on.
K+ deficiency has a far-reaching influence on agricultural. However, there are large numbers of K-deficient farmlands throughout the world. For example, 75% of the paddy soils in China are K deficient, as well as 67% of the wheat fields in Southern Australia3. Around the root zone, the concentration of soil potassium is generally less than 0.3 mM4. Thus most plants will face low-K+ stress in the process of growth. Most plants can resist low-K+ stress, mainly because they have set up strategy to acclimate to low-K+ stress. The main reason for plant adaption to low-K+ conditions is that they can keep a steady level of K+ in the cells and tissues via K+ transport5. Many high affinity potassium transporter genes, such as AtHAK5, HvHAK1 and OsHAK1, were induced by low K+ stress6,7,8. Recently, from monocotyledons, OsAKT19 and OsHAK510 have been functionally characterized in plant to demonstrate their key roles in K acquisition from low K supplied culture medium. What’s more, gene microarray analysis of Arabidopsis and rice identified that many candidate genes, including jasmonic acid-related enzymes, cell wall proteins, Ca2+ signaling proteins, protein kinase and ion transporter families, were induced by low K+ stress6,11,12.
Microarray technology, as a convenient tool, allows us to approach gene expression profiles of different plants in various external environments. For example, the transcriptome features in wheat responses to heat13, low temperature14, drought15 have already been detected by microarray technology. The results of microarray have indicated that the wheat transcriptome changes in responses to the above abiotic stress. Thus we can hypothesize that the gene expression levels of wheat may also change in response to K+-deficiency. Nevertheless, scarcely any development has been promoted in wheat transcriptional response to K+-deficient conditions. Wheat is a main grain crop, whose planting area, total output and total trade volume rank first in all types of crops. Therefore, it’s necessary to make clear the molecular mechanisms of wheat in response to K+-deficient conditions.
In the present study, Affymetrix GeneChip of wheat was used to investigating gene expression changes of wheat roots under K+-deficient conditions. In order to uncover the key genes in wheat adaptation to low-K+ stress, we selected two wheat genotypes which had different low-K+ tolerance for transcriptome analysis. “Tongzhou916” is tolerant to low K+ stress, whereas “Shiluan02-1” is susceptible to K+-deficiency. We investigated the function classification of differentially expressed genes and made a comparison of K+-deficiency responses between Tongzhou916 and Shiluan02-1. These results will provide a basis for our better understanding of molecular adaptation mechanisms in different wheat genotypes under K+-deficiency.
Results
Different sensitivity to K+ deficiency
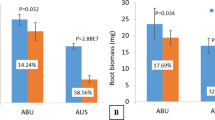
To investigate different genotype responses to K+ deficiency in wheat roots, we used two wheat cvs (K-effcient Tongzhou916, K-ineffcient Shiluan02-1) which had marked differences in sensitivity to K+ deficiency and root morphology. There were considerable phenotypic differences between Tongzhou916 and Shiluan02-1 during K+-sufficiency and K+-deficiency (CK and LK) for both two and three weeks (Fig. a,b). Though K+ deficiency inhibited growth of the two genotypes, Tongzhou916 had better shoot and root biomass under LK conditions (Fig. c,e), especially in three weeks. K efficiency coefficient was used to evaluate the K+-deficiency tolerance of the two wheat genotypes. The K efficiency coefficient of Tongzhou916 were considerably higher than that of Shiluan02-1 (Fig. 1d), indicating that Tongzhou916 had better tolerance to K+ deficiency. The differences in K efficiency coefficient between Tongzhou916 and Shiluan02-1 were more marked in three weeks. To make sure that the wheat seedlings were more sensitive to the changes in external K+ levels and the differences between the two wheat genotypes were more marked, we chose 3-week-old wheat seedlings, which required more potassium absorbed from the external environment and exhibited more considerable distinctions between the studied wheat genotypes.

Different sensitivity to K+ deficiency between two wheat cvs (K-efficient Tongzhou916 (TZ), K-inefficient Shiluan02-1(SL)).
Phenotypes of wheat seedlings during K+-sufficient (CK) and K+-deficient (LK) treatments for two weeks (a) and three weeks (b). Shoot biomass (c), K efficiency coefficient (d) and root biomass (e) of the two wheat genotypes under CK and LK conditions for two and three weeks. K efficiency coefficient (f), shoot biomass (g), root biomass (h), shoot K+ content (i) and root K+ content (j) of the two wheat genotypes at indicated times (after hydroponically grown in CK conditions for three weeks, wheat seedlings were transferred to CK and LK solutions for 1d, 3d, 5d and 7d, respectively). Data are means ± SE (n = 3). For (c), (d), (e) and (f) *, ** and ***denote significant differences at P < 0.05, 0.01 and 0.001, respectively. For (g), (h), (i) and (j), different letters denote significant differences at P < 0.05.
To study root transcriptional responses to K+ deficiency in Tongzhou916 and Shiluan02-1, firstly we need to ascertain the suitable period applied for the K+-deficient treatment. The K efficiency coefficient of Tongzhou916 was higher than that of Shiluan02-1 (Fig. 1f) at all times, indicating that Tongzhou916 had better tolerance to K+ deficiency. K efficiency coefficient began to decrease at 3 d. More remarkable differences between Tongzhou916 and Shiluan02-1 appeared at 5d. Moreover, the K+ content and biomass were both significantly decreased at 5d time point (Fig. 1g–j). Thus we used five days as the period of K+-deficient treatment to analyze variations in wheat root transcriptome during K+-deficient conditions.
Identification of differentially expressed genes in the two genotypes
The Wheat Genome Array used in this study is Affymetrix GeneChip® (Affymetrix, USA). In the present research, we selected two wheat genotypes to analyze the transcriptom changes during K+-deficient conditions. Since root is the first organ of plant which can detect element deficiency from the external environment, we harvested the root samples from K+-deficient and K+-sufficient treatments at 5d. We extracted total RNA of the harvested roots to carry out the following microarray experiments. In order to reduce the errors caused by the biological differences, we selected twenty wheat seedlings with uniform growth and collected roots of these seedlings to combine into one sample. In order to make sure that the microarray data is reproducible and reliable, we used three biological replications (each biological replication contained twenty individuals) for each experimental treatment. We detected over 61200 of the probe signals from Wheat Genome Array. To include more K+-deficiency responsive genes as possible, we used 1.5-fold (P < 0.05) for further analysis. To assess the reproducibility of microarray data, the correlation coefficients of biological replicates were calculated using GeneSpring GX 11 software. All correlation coefficients of each biological replications were greater than 0.94 (Supplementary Fig. S3A). From the cluster analysis, the microarrays of TZ-LK, TZ-CK, SL-LK and SL-CK clustered into one group, respectively (Supplementary Fig. S3B).
There were 2713 and 2485 differentially expressed genes (DEGs) in Tongzhou916 and Shiluan02-1, respectively (Table 1). Among these 2713 genes in Tongzhou916, there were 1321 up-regulated genes and 1392 down-regulated genes, up to 48.69% and 51.31% of the total DEGs, respectively. Among these 2485 genes in Shiluan02-1, 1177 genes were up regulated and 1308 genes were down regulated, accounting for 47.36% and 52.64% of the total DEGs, respectively. In both wheat genotypes, the number of down-regulated genes was in a slight advantage than that of up-regulated genes. This result was quite similar to the previous study, indicating that the expression levels of many genes in wheat might be inhibited to slow down the speed of wheat growth, whereas other genes were stimulated in order to acclimate to K+-deficient conditions12. In this study, there may be more up-regulated genes in Tongzhou916 than that in Shiluan02-1 at 5d, which may have an effect on the higher K+-deficiency tolerance in Tongzhou916 at 5d.
Hierarchical cluster method and Venn diagram were performed to study the commonness and individuality of transcriptome features in Tongzhou916 and Shiluan02-1 under different potassium treatments (Fig. 2). There were more up-regulated genes in Tongzhou916 than that in Shiluan02-1 under K+-deficient conditions. There were a certain number of genes up-regulated only in Tongzhou916 (Fig. 2a,b). The numbers of up-regulated and down-regulated genes in Tongzhou916 were 144 and 84 more than those in Shiluan02-1 (Fig. 2d,e). This suggested that more genes might be stimulated under 5-d K+-deficiency in Tongzhou916. In addition, there were 492 up-regulated genes and 519 down-regulated genes exhibited transcriptome changes in both wheat genotypes, which might make contributions to the response to K+-deficiency among the all wheat genotypes. From the hierarchical cluster of the shared DEG between the two genotypes (Fig. 2c), 70% of up-regulated genes had the higher expression levels in Tongzhou916 than those in Shiluan02-1, while less than 50% of down-regulated genes had the lower expression levels in Tongzhou916 than those in Shiluan02-1. In other words, most shared up-regulated genes showed higher expression levels in Tongzhou916 and over half the shared down-regulated genes displayed lower down-regulation degree in Tongzhou916.

Hierarchical cluster and Venn diagrams of differentially expressed genes in wheat response to K+-deficient conditions.
(a) Hierarchical cluster analysis of differentially expressed genes in Tongzhou916 (TZ) and Shiluan02-1(SL) during K+-sufficient (CK) and K+-deficient (LK) treatments. (b) Hierarchical cluster analysis of the differentially expressed genes (DEG) using averaged values of replicates in Tongzhou916 (TZ) and Shiluan02-1(SL) during K+ deficiency. (c) Hierarchical cluster analysis of the shared DEG using averaged values of replicates in TZ and SL during K+ deficiency. Venn diagrams of up-regulated (d) and down-regulated genes (e) under K+-deficient conditions.
Functional annotation of shared genes in two wheat genotypes response to K+ deficiency
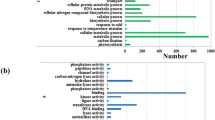
To classify differentially expressed genes of the two wheat genotypes according to the Gene Ontology (GO) terms, functional annotation was carried out. The 1011 shared genes, which showed significant changes under K+-deficient conditions in Tongzhou916 and Shiluan02-1 (P < 0.05), were divided into 14 main functional categories, according to the GO principles (Fig. 3a). Metabolic process (19%) was prominent among 14 main functional categories, followed by transport (7%), membrane (6%), cation binding (6%) and transferase activity (5%). Most of the above processes were thought to be associated with the response to K+-starvation closely. The GO analysis will provide the basis for us to better understand the transcriptome response to K+-deficiency in wheat roots.

Functional category distribution of differentially expressed genes shared in the two wheat genotypes under K+-deficient conditions.
(a) Percentages of differentially expressed genes in 14 main functional categories are shown. Detailed categories of differentially expressed genes in metabolic process (b) cation binding (c), transferase activity (d)
Through further analysis of the metabolic process category, we found that about 42% genes in this category were related to nitrogen compound metabolic process, 20% to carbohydrate metabolic process and 14% to phosphorus metabolic process (Fig. 3b). Many metabolic enzymes were involved in wheat response to K+-deficiency. Three probe sets encoding pyruvate decarboxylase isozyme were up-regulated under K+-deficient conditions (Table 2). Moreover, two probe sets (Ta.1870.1.S1_a_at and Ta.25990.1.A1_x_at) with putative functions in glutamate dehydrogenase were up-regulated, while one probe set (Ta.28435.1.S1_at) with putative functions in glutamate synthase was down-regulated under K+-deficiency. In addition, many other metabolic enzymes encoded genes, such as genes encoding sucrose synthase, ATPase and phosphatases were up-regulated. This suggested that genes encoding the regulation of metabolic enzymes might play a key role in wheat acclimation to K+-deficiency. For phosphorus metabolic process category, 24 genes encoding phosphorylation (11 up-regulated and 13 down-regulated) (Table 2), were transcriptionally regulated by K+-starvation.
In the category of cation binding, iron, zinc, magnesium and calcium binding proteins were included in the differentially expressed genes (Fig. 3c). There were 15 genes encoding peroxidase exhibited expressional changes in wheat response to K+-deficiency (including 8 down-regulated genes and 7 up-regulated genes), accounting for 71% of the iron-binding protein genes. We also identified 13 genes encoding calcium sensor protein, which showed transcriptional changes under K+-deficiency (5 up-regulated and 8 down-regulated) (Table 2).
In the transferase activity category, the main sub-category was kinase activity (45%) (Fig. 3d). Protein kinase as the main sub-category in kinase, is a kind of enzyme that catalyses protein phosphorylation. In this study, 31 genes encoding protein kinase were transcriptionally regulated (12 up-regulated and 19 down-regulated) under K+-deficiency (Table 2). The present result suggested that the process of phosphorylation and dephosphorylation might contribute a lot to the K+-deficiency management of wheat root. Methyltransferase is a kind of transferase, which can catalyze methyl into an acceptor molecule. In our microarray experiments, 11 genes encoding methyltransferase (5 up-regulated and 6 down-regulated) (Table 2), accounting for 12% of transferase activity category, showed changes in expression levels under K+-deficiency.
For ion transporters, three genes encoding nitrate transporter (Ta.5174.3.S1_x_at, Ta.8619.1.A1_at and Ta.21127.1.S1_at) were down-regulated after 5d of K+-starvation. In addition, the expression levels of two genes encoding peptide transporter (Ta.25793.1.S1_at and Ta.30027.1.S1_at) and one gene encoding ammonium transporter (Ta.27312.1.S1_x_at) also decreased under K+-deficient conditions (Table 2).
Functional annotation of specific K+-responsive genes in two wheat genotypes
In order to investigate the functions of specific K+-responsive genes in two wheat genotypes, GO analysis was carried out to compare the functional differences of specific up-regulated genes between two wheat genotypes. There were more specific up-regulated genes in Tongzhou916 (TZ) than those in Shiluan02-1 (SL) (Fig. 4), which suggested that more up-regulated genes had taken part in responding to K+-depletion in Tongzhou916. Although the main functions of two wheat genotypes were almost the same, the gene number of the main functions in Tongzhou916 was considerably higher than that in Shiluan02-1. Moreover, the function abundance of specific up-regulated gene in Tongzhou916 was higher than that in Shiluan02-1. There were five specific functions in Tongzhou916, including developmental process, cellular component organization, nutrient reservoir activity, antioxidant activity and membrane-enclosed lumen, which were not found in Shiluan02-1.

Numbers and Gene Ontology (GO) classifications of differentially expressed genes unique in the two wheat genotypes under K+-deficient conditions.
(a) Functional category distribution of differentially expressed genes unique in Tongzhou916 (TZ). (b) Functional category distribution of differentially expressed genes unique in Shiluan02-1(SL).
For metabolic process category, there were three genes with function in jasmonic acid biosynthesis (Ta.8990.1.S1_at, Ta.1207.1.S1_x_at and Ta.1207.1.S1_s_at) differentially expressed in Tongzhou916, while only one gene with function in jasmonic acid biosynthesis (Ta.20532.1.S1_at) in Shiluan02-1 (Table 3). In addition, there were three probe sets encoding vacuolar H+-pyrophosphatase up-regulated in Tongzhou916, while none probe set with such function showed changes in expression levels of Shiluan02-1. For catalytic activity, two probe sets encoding 1-aminocyclopropane-1-carboxylate oxidase (ethylene-related) were up regulated only in Tongzhou916. For response to stimulus category, we also found two jasmonic acid-related genes (Ta.8990.1.S1_at and Ta.27763.1.S1_at) in Tongzhou916, but none in Shiluan02-1. In addition, one ethylene-related probe set (Ta.8292.1.A1_at) and one probe set (Ta.25219.1.A1_at) encoding auxin response were found differentially expressed in Tongzhou916, while none was found differentially expressed in Shiluan02-1. Moreover, there were seven genes with functions in defense response (Ta.5720.1.S1_at, Ta.3828.2.S1_x_at, Ta.13907.2.S1_a_at, Ta.3590.1.S1_s_at, Ta.4328.1.S1_x_at, Ta.3467.2.S1_x_at and Ta.14281.1.S1_at) differentially expressed in Tongzhou916, while only one gene with functions in defense response (Ta.27229.1.S1_at) in Shiluan02-1 (Table 3). For transporter category, TaHKT1 and TaHAK2 genes were up-regulated in Tongzhou916, while only one high-affinity K+ transporter gene (TaHKT1) was up-regulated in Shiluan02-1.
For developmental process category, there were six genes with functions in anatomical structure development (Ta.6556.1.S1_x_at, Ta.1042.1.S1_x_at, Ta.6556.1.S1_at, Ta.1574.1.S1_s_at, Ta.449.1.S1_at and Ta.19563.1.S1_at) differentially expressed in Tongzhou916, none in Shiluan02-1 under K+-deficiency (Table 3). For nutrient reservoir activity category, which played a role in the storage of nutritious substrates, six related genes (Ta.9402.1.S1_x_at, Ta.722.1.A1_at, Ta.87.1.S1_at, Ta.25181.1.S1_at, Ta.87.1.S1_x_at and Ta.169.1.S1_x_at) differentially expressed in Tongzhou916, not in Shiluan02-1 under K+-deficiency. For antioxidant activity category, five peroxidase activity-related genes (Ta.8292.1.A1_at, Ta.22602.1.S1_a_at, Ta.21505.1.S1_at, Ta.22602.2.S1_x_at and Ta.21137.1.S1_x_at) among the specific genes were only differentially expressed in Tongzhou916 under K+-deficiency (Table 3).
Real-time fluorescence quantitative PCR (qRT-PCR) analysis
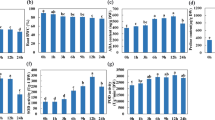
In order to verify the accuracy of microarray data, we chose ten genes randomly from the two wheat genotypes to perform qRT-PCR analysis. We designed specific-primers for the ten genes (see Supplementary Table S1). Quantitative variations of the selected genes between qRT-PCR and microarray were roughly similar (Fig. 5a), confirming that the results of microarray were reliable. In addition, we performed qRT-PCR analysis of the shared and specific genes mentioned in this paper. As shown in Fig. 5b, qRT-PCR results revealed that gene expression trends were significantly similar (r2 = 0.88) with those from the microarray data, indicating that our microarray results were reliable.

Verification of the transcriptome results through the experiments of qRT-PCR.
(a) Ten random genes were selected from Tongzhou916 (TZ) and Shiluan02-1(SL). The vertical axis represents quantitative variations of the selected genes, while horizontal axis represents probe names of the selected genes. Data are means ± SE (n = 3). (b) The relationships between qRT-PCR and microarray of the shared genes, specific genes and randomly selected genes. Values are the log2 ratio (K+-deficiency/K+-sufficiency) for genes. The determine coefficient (r2) is indicated in the figure. All qRT-PCR reactions were performed in three biological replicates.
Discussion
Valuable information about low K+-responsive genes has been shown above, which indicates that various physiological processes are involved commonly in wheat response to K+-deficiency and there are obvious transcriptional differences between the two wheat genotypes in response to K+-deficiency. Therefore, the Discussion section will focus on the following two questions: Which main physiological processes are involved commonly in wheat acclimation to K+-deficient conditions? Which special physiological processes or genes make Tongzhou916 more tolerant to low K+ stress?
Previous studies have emphasized the regulation role of some metabolic processes in plant adaptions to K+-deficiency11,12,16. The regulation of phosphorylation takes part in the modification of some transporters/channels for nutrient uptake. This mechanism can protect plants against the toxic damage caused by over accumulation, or provide energy for acquisition rate if needed17. Lee et al.18 reported that some proteins together with protein phosphatases made up the phosphorylation and dephosphorylation of protein, which played a key role in the K+ transport regulation. In the present data, several genes encoding phosphatase and phosphorylation were transcriptionally regulated, which indicated that phosphorylation and dephosphorylation also contributed a lot to wheat adaption to K+-deficient conditions. According to the previous study, the activity of OsAKT1 (as well as AtAKT1) is phosphorylated upon K deficiency9. In response to a signal event of K+-deficiency, phosphorylation may stimulates some K+ channels in wheat root, while this signal incident may be momentary and restrained by a signal terminator, which will lead to dephosphorylating, namely deactivation and recovery of K+ channels to the original state.
Metabolic enzymes contribute a lot to Arabidopsis acclimation to K+-deficient conditions16. Besides genes encoding phosphatase, several genes encoding other metabolic enzymes (such as ATPase, glutamate-related enzyme, sucrose synthase and pyruvate decarboxylase isozyme) had transcription regulation in wheat response to K+-starvation. ATPase activity is stimulated directly by external K19. Potassium channels together with the proton ATPase dominate the electric conductance of the plasma membrane and, therefore, their relative conductance determines the membrane potential, affecting the driving force for K+ movement in turn20. According to the present data, four genes encoding ATPase were up-regulated under K+-deficiency, which could lead to the increase of ATPase in wheat root. ATPase can improve the ATP hydrolysis and energy release, which can provide a driving force for transmembrane transport of potassium. Thus the up-regulations of genes encoding ATPase may improve K+-deficiency tolerance in wheat root. Glutamate may directly regulate ion channels of the putative glutamate receptor family20. Glutamate may affect cation transportation probably because that glutamate can cause large membrane depolarization of root cells and a change of cytosolic Ca2+ concentration21. In this study, two genes with putative functions in glutamate dehydrogenase were up-regulated, while one gene with putative functions in glutamate synthase was down-regulated under K+-deficiency. These glutamate-related enzyme genes can lead to the decrease of glutamate in wheat root, which result is similar to the study on K+-starved Arabidopsis20. Thus we can hypothesize that glutamate may involve in K+-deficiency signaling of wheat root cells under K+-deficient conditions. Many other metabolic enzymes showed marked changes when plants were in the face of K+-deficient conditions, these may be due to the K+-induced transcriptional variations in genes encoding metabolic enzymes.
A previous research indicated that peroxidases (heme-containing proteins) contributed a lot to Arabidopsis response to K+-deficient conditions11. One reason for this was likely that peroxidases were involved in cell wall responses associated with growth. Another reason for the participation of peroxidases in adaption to K+-starvation was likely to be their contributions to ROS detoxification, for H2O2 played a role of signal in K stress sensing22. Among the shared genes, 15 genes encoding peroxidase, accounting for more than two-thirds of the iron ion binding genes, showed changes in transcription under K+-deficiency. These genes may participate in growth-related cell wall responses and ROS detoxification of wheat root and thus make contributions to wheat acclimatization to K+-starvation. Among the unique genes, several peroxidase-related genes were only found up-regulated in Tongzhou916, which may take a positive part in root growth and ROS production. Thus, the up-regulations of peroxidase-related genes may make a great contribution to promoting low-K+ tolerance of Tongzhou916. Previous studies found large proportion of Ca2+-binding proteins among K-regulated transcripts, which strongly suggested that intracellular Ca, to some extent, participated in responding to K+-deficiency11,12. In our microarray data, 13 calcium sensor protein genes were also found participating in wheat response to K+-deficient conditions, indicating that these genes might link calcium signaling and downstream target proteins in wheat adaption to K+-starvation.
Previous researchers found that many protein kinase genes differentially expressed under ionic stress23,24. Protein kinases regulated the transportation of plant K+, which was the fact revealed by some previous studies25,26,27. In the present data, a number of genes encoding protein kinase exhibited variations in expression levels of wheat under K+-deficient conditions. So we hypothesized that protein kinase encoding genes, which showed expression changes under K+-deficiency, might participated in wheat adaption to K+-starvation. These protein kinases were likely to perceive external low-K+ condition and regulate ion transport through phosphorylating ion transporters in plant cells and thus enhanced transporter-mediated K uptake in wheat roots. Armengaud et al.11 found that the expression levels of some methyltransferase or methyltransferase-like proteins genes showed changes under K+-deficient and K+-resupplied conditions. Transfer of methyl groups via S-adenosyl-methyltransferase appears in some hormonal pathways (e.g. JA pathway). We also found several genes encoding methyltransferase showed changes in expression levels under K+-deficiency. These genes may affect the transfer of methyl groups and thus influence some K+-related hormonal pathways. So we can hypothesize that methyltransferase maybe another low K+-responsive transferase.
Previous researches on Arabidopsis and rice reported that several nitrate transporter genes were down-regulated during K+-deficiency11,12. Interestingly, several genes encoding peptide transporters also took part in the above response11. By contrast, the lack of N made the expression levels of potassium transporter genes increase28. Moreover, a similar relationship between NH4+ and K+ was found in barley and Arabidopsis29,30. In the present research, some genes encoding nitrate, peptide and ammonium transporters were down-regulated upon K+ starvation, which suggested some subtle relationship between K+ uptake and N transport. AtNRT1.5 mRNA levels were down-regulated when potassium was limiting, suggesting that root-to-shoot nitrate transport is controlled by potassium levels31. Xia et al.32 found that OsNPF2.4 functions in acquisition and long-distance transport of NO3− and that altering its expression has an indirect effect on K recycling between the root and shoot. These findings, suggesting that nitrogen metabolites were likely to take part in the signaling of low-K+ stress, provide a basis for molecular research on the relationship between potassium and nitrogen. Various high-affinity K+-transporter genes (such as OsHAK1, HvHAK1, TaHKT1 and AtHAK5), derived from different plants, are stimulated by K+-deficient treatment6,7,8,33. Moreover, from monocotyledons, OsAKT19 and OsHAK510 have been functionally characterized in plant to demonstrate their key roles in K acquisition from low K supplied culture medium. It may be a direct and effective strategy that K+ transporter genes will up-regulate in overcoming K+-deficient conditions. According to the present data, TaHKT1 was up-regulated in both wheat genotypes. In addition, TaHAK2 (a member of the KUP/HAK/KT family) was only up-regulated in Tongzhou916 under K+-starvation. Haro et al.34 reported that PpHAK2 might encode a K+/H+ antiporter or a K+-H+ symporter, which regulated the H+ transportation between endoplasmic reticulum lumen and cytosol. Under K+-starvation, the increased expression level of TaHAK2 may contribute to the strong low K+-tolerance of Tongzhou916.
Several jasmonic acid-related genes were found Arabidopsis root responding to K+-deficiency and K+-resupply11. Our data showed that jasmonic acid-related genes in Tongzhou916 were more abundant than those in Shiluan02-1. Jasmonic acid contributes a lot to the potassium signaling and management via various physiological and metabolic processes under low K+-stress. For example, jasmonic acid may make plants adapt to low-K+ stress through nutrient storage and remobilization11. Jasmonic acid may also reduce the pathogen attack in K-starved plants, which can also make decrease in damages from K+ starvation20. Therefore, more jasmonic acid-related genes in Tongzhou916 can make better low-K+ stress acclimation for Tongzhou916.
V-PPase can connect the H+-transmembrane transport and free energy generated by hydrolysis process of inorganic pyrophosphate (PPi), which makes V-PPase more predominant in the young and developing cells35. In additon, V-PPase can build the transmembrane electrochemical gradients, providing a driving force for the active transmembrane movement of various solutes (e.g. cation)36. Moreover, a previous study reported that V-PPase also took part in the transmembrane transportation of potassium37. In this study, shared genes encoding V-PPase were only up-regulated in Tongzhou916, which might improve the root K+ uptake in Tongzhou916. In addition, Arabidopsis V-PPase (AVP1) promotes the flow of auxin, which regulates the growth of root system38. The root of Tongzhou916 was better developed than that of Shiluan02-1 under low-K+ stress (Supplementary Table S2). Therefore, we can hypothesize that these genes encoding V-PPase may involve in the regulation of root development and root K+ uptake in Tongzhou916 and thus enhance the low-K+ tolerance of Tongzhou916.
Ethylene makes a great contribution to the development of root system39. Previous microarray data suggested that genes encoding ethylene showed changes in Arabidopsis response to low-K+ stress22. Ethylene also stimulated the development of primary root and root hair, which enhanced the Arabidopsis tolerance to K+-deficient conditions40. In the present data, shared genes encoding 1-aminocyclopropane-1-carboxylate oxidase, which took part in biosynthesis of ethylene, were only up-regulated in Tongzhou916. Ethylene responsive gene also showed changes only in Tongzhou916 after 5d K+-starvation. Auxin is important for root architecture41. Among the shared genes, IAA9-auxin-responsive gene, an IAA family member, only showed increase expression levels in Tongzhou916. For root architecture can be modified by the interactions between ethylene and auxin, ethylene-related and auxin-related genes may contribute a lot to the difference of root architecture between Tongzhou916 and Shiluan02-1 (Supplementary Table S2). Better root development made Tongzhou916 more tolerant to low-K+ stress.
For phosphorus and nitrogen starvation, several defense-related genes were transcriptionally regulated in Arabidopsis42,43. Some plant defensin genes were also found transcriptional changes in Arabidopsis under K+-starvation. The increase jasmonic acid in K+-starved plants will have certain effects on inducing defense response11. Defense response can prevent plants from insect and pathogen damages, which enemies K+-starved plants are likely to face44,45. Our microarray data showed that there were more genes encoding defense response remarkably up-regulated in Tongzhou916 under K+-deficient conditions, which maybe an important factor protecting Tongzhou916 under long-term K+-starvation.
Anatomical structure traits refer to the arrangement of the tissues and cells and the characteristics of plant internal structure. Root anatomical structure, such as cortical size, the number and arrangement of root cells, determine the pathway and rate of nutrient uptake from the soil solutes to the root vascular cylinder46. Postma and Lynch47 reported that small changes in root anatomical structure were likely to affect physiology and growth of the whole plant, which had possible value for K+-uptake. From our microarray data, there were six genes encoding anatomical structure development showed transcriptionally changes in Tongzhou916, while none was in Shiluan02-1. These findings suggested that Tongzhou916 might adapt itself to K+-starvation through anatomical structure development.
Plants accumulate nutrients in the form of polysaccharides and proteins in appropriate organs and assimilate them into a wide variety of compounds for their growth and organogenesis. Nutrient reservoir (storage protein) is important for the storage of nutritious substrates. The existence of nutrient reservoir not only prevents the loss of nutrients, but also provides necessary nutrient for plants in the new growing season48. Armengaud et al.11 reported that several genes encoding vegetative storage protein took part in Arabidopsis response to external K+-levels. In this study, several genes encoding nutrient reservoir were up-regulated in Tongzhou916 after 5d K+-starvation, while none gene with such function was up-regulated in Shiluan02-1. This suggested that Tongzhou916 could better prevent nutrient loss via nutrient reservoirs during K+-starvation and thus made Tongzhou916 more tolerant to low-K+ stress.
In conclusion, these two wheat genotypes exhibited variant transcription features under K+-deficiency. There were more up-regulated genes involved in the response of low-K+ tolerant genotype to K+ starvation. The identified genes that responded to K+-deficiency belonged to metabolic process, cation binding, transferase activity, ion transporters and so forth. A lot of genes associated with jasmonic acid and defense response were considerably up-regulated, while the numbers of genes related to these processes in low-K+ tolerant genotype were higher than those in low-K+ susceptible genotype. In addition, some unique genes, such as vacuolar H+-pyrophosphatase, ethylene-related, auxin response, anatomical structure development and nutrient reservoir genes, were involved in low-K+ tolerant genotype. These unique genes played a key role in root architecture, K+ uptake and nutrient storage, which might contribute a lot to the strong low-K+ tolerance in wheat. However, further research is required to make it clear that how these specific genes regulate potassium metabolism.
Methods
Plant materials and low-K+ stress treatment
Two wheat genotypes, low-K+ tolerant “Tongzhou916” and low-K+ susceptible “Shiluan02-1” were used in this study. Two wheat genotypes were cultured hydroponically in an artificial climatic box. Environmental conditions of this box were as follows: 16/8 h day/night regime, 25/18 °C temperature, 70% atmospheric humidity and 30000 lux illumination intensity in the daytime. The normal nutrient solution was as follows (mmol L−1): Ca(NO3)2•4H2O 1.0, MgSO4•H2O 1.0, NaH2PO4 0.25, NH4NO3 1.0, CaCl2 1.5, Fe-EDTA 0.1, MnSO4•H2O 1.0 × 10−3, ZnSO4•7H2O 1.0 × 10−3, CuSO4•5H2O 5.0 × 10−4, (NH4)6 Mo7O24•4H2O 5.0 × 10−6, H3BO4 1.0 × 10−3 and K2SO4 1.0, (pH 6.5). Nutrient solution was ventilated 12 h by pumps each day and replaced every day.
In the first experiment, K+-sensitivities of the two wheat genotypes were expected to be validated under low-K+ conditions. Two wheat genotypes were grown in normal and low-K+ conditions for two or three weeks, respectively. In this experiment, the concentration of K+ was 0.01 mM (K2SO4 5.0 × 10−3 mmol L−1) in low-K+ conditions. Two wheat genotypes were harvested after two and three weeks. The growth states and K-sensitivity of the two wheat genotypes were then investigated. In the second experiment, we want to get the optimum time point of K+-deficient treatment for investigating the transcriptome differences in wheat roots during K+-starvation. In this experiment, wheat seedlings were firstly grown in normal nutrition solutions for 3 wk. Secondly, half of the above seedlings were transplanted to nutrient solution without K2SO4 (i.e. K+-deficient treatment (LK)), while another half were transplanted to nutrient solution with K2SO4 as control (i.e. control treatment (CK)). Wheat seedlings of the above two treatments were collected at 1, 3, 5 and 7 d for K-sensitivity assays. In the last experiment, the growth conditions were the same as that in the second experiment. Wheat roots of CK and LK were collected at 5d and quickly put into liquid nitrogen for following RNA isolation.
Microarray analysis
The GeneChip experiments were finished with the assist of Shanghai Biotechnology Corporation. Total RNA was extracted using TRIZOL reagent (Cat#15596-018, Life technologies, Carlsbad, CA, US) following the manufacturer’s instructions and checked for a RIN number to inspect RNA integrity by an Agilent Bioanalyzer 2100 (Agilent technologies, Santa Clara, CA, US). Qualified total RNA was further purified by RNeasy micro kit (Cat#74004, QIAGEN, GmBH, Germany) and RNase-Free DNase Set (Cat#79254, QIAGEN, GmBH, Germany).
Total RNA were amplified, labeled and purified by using GeneChip 3’IVT Express Kit (Cat#901229, Affymetrix, Santa Clara, CA, US) followed the manufacturer’s instructions to obtain biotin labeled cRNA49. Array hybridization and wash was performed using GeneChip® Hybridization, Wash and Stain Kit (Cat#900720, Affymetrix, Santa Clara, CA, US) in Hybridization Oven 645 (Cat#00-0331-220 V, Affymetrix, Santa Clara, CA, US) and Fluidics Station 450 (Cat#00-0079, Affymetrix, Santa Clara, CA, US) followed the manufacturer’s instructions. Slides were scanned by GeneChip® Scanner 3000 (Cat#00-00212, Affymetrix, Santa Clara, CA, US) and Command Console Software 3.1 (Affymetrix, Santa Clara, CA, US) with default settings50.
qRT-PCR Analysis
Part of the wheat roots, which were applied to perform gene chip detection, was applied for the qRT-PCR determination to verify the microarray results. TRIZOL reagent (Cat#15596-018, Life technologies, Carlsbad, CA, US) was used to extract total RNA of the root samples. The actin gene of Triticum aestivum L. was selected as the endogenous control. The extracted total RNA was applied to synthesize first-strand cDNA by cDNA synthesis Kit (Promega, USA) according to the manufacturer’s instructions. Three biological replicates (each biological replication contained twenty individuals) were performed in this experiment. qRT-PCR was performed using a 20 μl reaction system, containing 10 μl of 2X-RTmix, 2 μl template (0.2 μM), 1 μl forward primer, 1 μl reverse primer and 6 μl nuclease-free water. PCR primers in this experiment (Supplementary Table S1) were designed by Primer 5 and DNAMAN software. Each PCR experiment was repeated at least for three times. The relative quantitative method of ΔΔ CT was applied to analyze the quantitative changes of the selected genes in the two treatments51.
Biomass and K+ content measurement
The shoots and roots of different treatments were harvested at various times. The shoots and roots were collected separately, dried at 80 °C for 48 h and then weighed (dry weight). K content in plant was determined by the method as described by Mills and Jones52, which involved digesting plant in a mixture of H2SO4 and H2O2. Potassium concentration in the digested solution was determined by a flame photometer. Three biological replicates were used for biomass and K+ content measurements. The K efficiency coefficient was calculated as follows53:
K efficiency coefficient = shoot dry weight under LK/shoot dry weight under CK (1)
Statistical analysis
MAS 5.0 algorithm, Gene Spring Software 11.0 (Agilent technologies, Santa Clara, CA, US) was used to realize the normalization of data generated by the scanner. Only genes showing transcriptional differences (>1.5 fold-change and P < 0.05) were screened for further analysis. A positive or negative value represents up or down regulation, respectively. SBC Analysis System ( http://www.sas.ebioservice.com) and AgriGO online service ( http://bioinfo.cau.edu.cn/agriGO) were applied for the data analysis and functional annotation. T-test and ANOVA were carried out to test the significance of the type differences. For statistical analysis of data SPSS window version 17 (SPSS Inc., Chicago, USA) and Microsoft Excel (Microsoft Corporation, USA) were used. OriginPro 8.1 (Origin Inc., Chicago, USA) was used to draw the figures.
Additional Information
How to cite this article: Ruan, L. et al. Comparative analysis of potassium deficiency-responsive transcriptomes in low potassium susceptible and tolerant wheat (Triticum aestivum L.). Sci. Rep. 5, 10090; doi: 10.1038/srep10090 (2015).
References
Leigh, R. A. & Wyn Jones, R. G. A hypothesis relating critical potassium concentrations for growth to the distribution and function of this ion in the plant cell. New Phytol. 97, 1–13 (1984).
Römheld, V. & Kirkby, E. A. Research on potassium in agriculture: needs and prospects. Plant Soil. 335, 155–180 (2010).
Rengel, Z. & Damon, P. M. Crops and genotypes differ in efficiency of potassium uptake and use. Physiol. Plantarum 133, 624–636 (2008).
Mengel, K. & Kirkby, E. A. in Principles of Plant Nutrition 3rd edn, Vol. 1 (eds Kosegarten, H. et al. ) Ch. 10, 655 International Potash Institute, Worblaufen-Bern, Switzerland, 1982).
Amtmann, A., Armengaud, P. & Volkov, V. in Membrane Transport in Plants 1st edn, Vol. 15 (ed Michael, R. B. ) Ch. 2, 25–36 Blackwell Publishing: Oxford, 2004).
Gierth, M., Maser, P. & Schroeder, J. I. The potassium transporter AtHAK5 functions in K+ deprivation-induced high-affinity K+ uptake and AKT1 K+ channel contribution to K+ uptake kinetics in Arabidopsis roots. Plant Physiol. 137, 1105–1114 (2005).
Fulgenzi, F. R. et al. The ionic environment controls the contribution of the barley HvHAK1 transporter to potassium acquisition. Plant Physiol. 147, 252–262 (2008).
Bañuelos, M. A., Garciadeblas, B., Cubero, B. & Rodríguez-Navarro, A. Inventory and functional characterization of the HAK potassium transporters of rice. Plant Physiol. 130, 784–795 (2002).
Li, J. et al. The Os-AKT1 channel is critical for K+ uptake in rice roots and is modulated by the rice CBL1–CIPK23 complex. Plant Cell. 26, 3387–3402 (2014).
Yang, T. et al. The role of OsHAK5 in potassium acquisition and transport from roots to shoots in rice at low potassium supply levels. Plant Physiol. 166, 945–959 (2014).
Armengaud, P., Breitling, R. & Amtmann, A. The potassium-dependent transcriptome of Arabidopsis reveals a prominent role of jasmonic acid in nutrient signaling. Plant Physiol. 136, 2556–2576 (2004).
Ma, T. L., Wu, W. H. & Wang, Y. Transcriptome analysis of rice root responses to potassium deficiency. BMC Plant Biol. 12, 161 (2012).
Qin, D. D. et al. Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using Wheat Genome Array. BMC Genomics 9, 432 (2008).
Gulick, P. J. et al. Transcriptome comparison of winter and spring wheat responding to low temperature. Genome 48, 913–923 (2005).
Krugman, T. et al. Multilevel regulation and signalling processes associated with adaptation to terminal drought in wild emmer wheat. Funct. Integr. Genomic . 10, 167–186 (2010).
Armengaud, P. et al. Multilevel analysis of primary metabolism provides new insights into the role of potassium nutrition for glycolysis and nitrogen assimilation in Arabidopsis roots. Plant Physiol. 150, 772–785 (2009).
Ho, C. H. & Tasy, Y. F. Nitrate, ammonium and potassium sensing and signaling. Curr. Opin. Plant Biol. 13, 604–610 (2010).
Lee, S. C. et al. A protein phosphorylation/dephosphorylation network regulates a plant potassium channel. Proc. Natl. Acad. Sci. U.S.A. 104, 15959–15964 (2007).
Hall, J. L. & Williams, L. E. Properties and functions of proton pumps in higher plants. Pestic. Sci. 32, 339–351 (1991).
Amtmann, A., Hammond, J. P., Armengaud, P. & White, P. J. Nutrient sensing and signalling in plants: potassium and phosphorus. Adv. Bot. Res. 43, 209–256 (2006).
Dennison, K. L. & Spalding, E. P. Glutamate-gated calcium fluxes in Arabidopsis. Plant Physiol. 124, 1511–1514 (2000).
Shin, R. & Schachtman, D. P. Hydrogen peroxide mediates plant root cell response to nutrient deprivation. Proc. Natl. Acad. Sci. U.S.A. 101, 8827–8832 (2004).
Weinl, S. & Kudla, J. The CBL-CIPK Ca2+-decoding signaling network: function and perspectives. New Phytol. 184, 517–528 (2009).
McCormick, J. A. & Ellison, D. H. The WNKs: atypical protein kinases with pleiotropic actions. Physiol. Rev. 91, 177–219 (2011).
Held, K. et al. Calcium dependent modulation and plasma membrane targeting of the AKT2 potassium channel by the CBL4/CIPK6 calcium sensor/protein kinase complex. Cell Res. 21, 1116–1130 (2011).
Hong-Hermesdorfa, A., Brüxa, A., Grüberb, A., Grüberb, G. & Schumacher, K. A WNK kinase binds and phosphorylates V-ATPase subunit C. FEBS Lett. 580, 932–939 (2006).
Xu, J. et al. A protein kinase, interacting with two calcineurin B-like proteins, regulates K+ transporter AKT1 in Arabidopsis. Cell 125, 1347–1360 (2006).
Shin, R., Berg, R. H. & Schachtman, D. P. Reactive oxygen species and root hairs in Arabidopsis root response to nitrogen, phosphorus and potassium deficiency. Plant Cell Physiol. 46, 1350–1357 (2005).
Santa-Maria, G. E., Danna, C. H. & Czibener, C. High-affinity potassium transport in barley roots: Ammonium-sensitive and-insensitive pathways. Plant Physiol. 123, 297–306 (2000).
Maathuis, F. J. M. et al. Transcriptome analysis of root transporters reveals participation of multiple gene families in the response to cation stress. Plant J. 35, 675–692 (2003).
Lin, S. H. et al. Mutation of the Arabidopsis NRT1.5 nitrate transporter causes defective root-to-shoot nitrate transport. Plant Cell 20, 2514–2528 (2008).
Xia, X. D. et al. Rice nitrate transporter OsNPF2.4 functions in low-affinity acquisition and long-distance transport. J. Exp. Bot. 66, 317–331 (2015).
Garciadeblás, B., Senn, M. E., Bañuelos, M. A. & Rodríguez-Navarro, A. Sodium transport and HKT transporters: the rice model. Plant J. 34, 788–801 (2003).
Haro, R., Fraile-Escanciano, A., Gonzalez-Melendi, P. & Rodriguez-Navarro, A. The potassium transporters HAK2 and HAK3 localize to endomembranes in Physcomitrella patens. HAK2 is required in some stress conditions. Plant Cell Physiol. 54, 1441–1454 (2013).
Nakanishi, Y. & Maeshima, M. Molecular cloning of vacuolar H+-pyrophosphatase and its developmental expression in growing hypocotyl of mung bean. Plant Physiol. 116, 589–597 (1998).
Mitsuda, N. et al. Novel type Arabidopsis thaliana H+-PPase is localized to the Golgi apparatus. FEBS Lett. 488, 29–31 (2001).
Obermeyer, G., Sommer, A. & Bentrup, F. W. Potassium and voltage dependence of the inorganic pyrophosphatase of intact vacuoles from Chenopodium rubrurn. BBA. Biomembranes 1284, 203–212 (1996).
Li, J. S. et al. Arabidopsis H+-PPase AVP1 regulates auxin-mediated organ development. Science 310, 121–125 (2005).
He, Z., Ma, Z., Brown, K. M. & Lynch, J. P. Assessment of in equality of root hair density in Arabidopsis thaliana using the Gini coeffcient: A close look at the effect of phosphorus and its interaction with ethylene. Ann. Bot. 95, 287–293 (2005).
Jung, J. Y., Shin, R. & Schachtman, D. P. Ethylene mediates response and tolerance to potassium deprivation in Arabidopsis. Plant Cell 21, 607–621 (2009).
Al-Ghazi, Y. et al. Temporal responses of Arabidopsis root architecture to phosphate starvation: evidence for the involvement of auxin signalling. Plant Cell Environ. 26, 1053–1066 (2003).
Wu, P. et al. Phosphate starvation triggers distinct alterations of genome expression in Arabidopsis roots and leaves. Plant Physiol. 132, 1260–1271 (2003).
Wang, R., Okamoto, M., Xing, X. & Crawford, N. M. Microarray analysis of the nitrate response in Arabidopsis roots and shoots reveals over 1,000 rapidly responding genes and new linkages to glucose, trehalose-6-phosphate, iron and sulfate metabolism. Plant Physiol. 132, 556–567 (2003).
Kessler, A. & Baldwin, I. T. Plant responses to insect herbivory: the emerging molecular analysis. Annu. Rev. Plant Biol. 53, 299–328 (2002).
Kunkel, B. N. & Brooks, D. M. Cross talk between signaling pathways in pathogen defense. Curr. Opin. Plant Biol. 5, 325–331 (2002).
Burton, A. L. Phenotypic evaluation and genetic basis of anatomical and architectural root traits in the genus Zea . PhD thesis. The Pennsylvania State University: University Park, PA, ), pp. 11–13 (2010).
Postma, J. A. & Lynch, J. P. Root cortical aerenchyma enhances the growth of maize on soils with suboptimal availability of nitrogen, phosphorus and potassium. Plant Physiol. 156, 1190–1201 (2011).
Utsugi, S., Sakamoto, W., Murata, M. & Motoyoshi, F. Arabidopsis thaliana vegetative storage protein (VSP) genes: gene organization and tissue-specific expression. Plant Mol. Biol. 38, 565–576 (1998).
Wu, L. H., Wang, Y., Nie, J., Fan, X. H., Cheng, Y. Y. A network pharmacology approach to evaluating the efficacy of Chinese medicine using genome-wide transcriptional expression data. Evid-Based. Compl. Alt . 2013, 1–7 (2013).
Zeng, Q. Q. et al. Quantitative proteomics reveals ER-α involvement in CD146-induced epithelial-mesenchymal transition in breast cancer cells. J. Proteomics 103, 153–169 (2014).
Revel, A. T., Talaat, A. M. & Norgard, M. V. DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. Proc. Natl. Acad. Sci. U.S.A. 99, 1562–1567 (2002).
Mills, H. A. & Jones, J. B. in Plant analysis handbook 2nd edn, Vol. 2 (eds Wolf, B. et al. ) Ch. 3, 45–48 Micro-Macro: Athens, 1996).
Mengel, K. in Methods of K-Research in Plants Proc. 21st Colloq. Intern. 1st edn, Vol. 1 (eds Wittchen, H. U. et al. ) Ch. 5, 67–76 Potash Institute, Israel 1989).
Acknowledgements
This study was funded by the National Basic Research Program of China (No. 2011CB100506), China Agriculture Research System-Wheat (CARS-03-1-28), Science and Technology Service Network Initiative (KFJ-EW-STS-055). We are grateful to Professor Anthony J. Miller for his help in technical guidance and language modification. We would also like to thank Shanghai Biotechnology Corporation for their helpful technical support.
Author information
Authors and Affiliations
Contributions
J.B.Z. and L.R. designed the experiment. L.R. conducted the measurements, data analysis and wrote the manuscript. X.L.X., C.Z.Z. and D.H.M. assisted with the data analysis. L.C. and B.Z.Z. assisted with the experiment. These authors reviewed the manuscript before the submission.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ruan, L., Zhang, J., Xin, X. et al. Comparative analysis of potassium deficiency-responsive transcriptomes in low potassium susceptible and tolerant wheat (Triticum aestivum L.). Sci Rep 5, 10090 (2015). https://doi.org/10.1038/srep10090
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10090
This article is cited by
-
Comparative physiological and transcriptome analysis between potassium-deficiency tolerant and sensitive sweetpotato genotypes in response to potassium-deficiency stress
BMC Genomics (2024)
-
Dynamic transcriptome analysis unravels key regulatory genes of maize root growth and development in response to potassium deficiency
Planta (2023)
-
Plasticity of wheat seedling responses to K+ deficiency highlighted by integrated phenotyping of roots and root hairs over the whole root system
Stress Biology (2023)
-
Transcriptome analysis of maize seedling roots in response to nitrogen-, phosphorus-, and potassium deficiency
Plant and Soil (2020)
-
Physiology and proteomic analysis reveals root, stem and leaf responses to potassium deficiency stress in alligator weed
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
