
Abstract
Rex rabbit is an important small herbivore for fur and meat production. However, little is known about the gut microbiota in rex rabbit, especially regarding their relationship with different fecal types and growth of the hosts. We characterized the microbiota of both hard and soft feces from rex rabbits with high and low body weight by using the Illumina MiSeq platform targeting the V4 region of the 16S rDNA. High weight rex rabbits possess distinctive microbiota in hard feces, but not in soft feces, from the low weight group. We detected the overrepresentation of several genera such as YS2/Cyanobacteria and Bacteroidales and underrepresentation of genera such as Anaeroplasma spp. and Clostridiaceae in high weight hard feces. Between fecal types, several bacterial taxa such as Ruminococcaceae, and Akkermansia spp. were enriched in soft feces. PICRUSt analysis revealed that metabolic pathways such as “stilbenoid, diarylheptanoid, gingerol biosynthesis” were enriched in high weight rabbits and pathways related to “xenobiotics biodegradation” and “various types of N-glycan biosynthesis” were overrepresented in rabbit soft feces. Our study provides foundation to generate hypothesis aiming to test the roles that different bacterial taxa play in the growth and caecotrophy of rex rabbits.
Similar content being viewed by others

Introduction
Rex rabbit is an important small herbivorous mammal widely raised for fur and meat production. Rex rabbit excretes two types of feces: hard feces which contain poorly digestible large particles and soft feces which consist of fermented soft fine particles from caecum fermentation1,2. As a caecotrophic small animal, rex rabbit eats soft feces, which provides additional proteins, vitamins and inorganic salt. Earlier studies have shown the differences in nutrients between hard and soft feces3,4,5. However, little is known about the composition of the microbiota of soft and hard feces6,7.
Gut microbiota play important roles in mammal's health and production. Studies have shown that gut microbiota are associated with many key functions of the host, such as obesity8,9, gut immune maturation10 and nutrition restriction11. We thus hypothesize that the gut microbiota differs in fecal types and is also associated with the growth of rex rabbit.
The objectives of this study were: i) to characterize and compare the microbiota in hard and soft feces in rex rabbits and ii) to identify bacterial taxa that are associated with the growth of rex rabbits.
Methods
Experimental design and sampling
Our animal experiment was approved by the Institutional Animal Care and Use Committee of the Sichuan Agricultural University under permit number DKY- S20123122 and was performed in the breeding center of rex rabbit research institution located in the suburb of Xinjin County, Chengdu, China. All rex rabbits were fed with customized fodder (probiotics and antibiotics free) and raised under the same temperature (25 ± 1.5°C controlled by automatic heating and ventilation devices). Figure S1 shows the flowchart of this study. Briefly, 80 young female rex rabbits (breed Sichuan White rex rabbit) born on the same day from different rabbit mothers, were raised in separated cages after weaning (day 40) to minimize the confounding effects of genetics and families. Their body weights were sorted on day 70 and the top 10 rex rabbits with the highest weight (HW) and the bottom 10 with the lowest weight (LW) were selected in this study. On day 90, their body weights were measured again and both hard and soft fecal samples were collected.
All experiments were performed in accordance with the approved guidelines and regulations.
For soft fecal sample collection, all rabbits were forced to wear the caecotrophy prevention circle (CP circle, Figure S2) for 10 hours (from 22:00 pm on the day before sampling to 8:00 am on the sampling day) to prevent their caecotrophic behavior. Both the hard and soft feces were automatically dropped into the collection tray under the cage. Fresh fecal samples were immediately transferred into liquid nitrogen container for temporary storage before they were sent to the laboratory where the samples were stored at −80°C.
DNA extraction and pyrosequencing
Total bacteria DNA was extracted from fecal samples by using PowerFecal™ DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) according to manufacturer's instruction and was stored at −80°C before further analysis. Sequencing was performed at the Novogene Bioinformatics Technology Co., Ltd. Briefly, DNA was amplified by using the 515f/806r primer set (515f: 5′-GTG CCA GCM GCC GCG GTA A-3′, 806r: 5′-XXX XXX GGA CTA CHV GGG TWT CTA AT-3′), which targets the V4 region of the bacterial 16S rDNA, with the reverse primer containing a 6-bp error-correcting barcode unique to each sample. PCR reaction was performed using phusion high-fidelity PCR Mastermix (New England Biolabs (Beijing) LTD., China) with the following condition: 94°C for 3 min (1 cycle), 94°C for 45 s/50°C for 60 s/72°C for 90 s (35 cycles) and a last step of 72°C for 10 min. PCR products were purified by using the QIAquick Gel Extraction Kit (QIAGEN, Dusseldorf, Germany). Pyrosequencing was conducted on an Illumina MiSeq 2 × 250 platform according to protocols described by Caporaso, et al12.
Bioinformatics and statistical analysis
Sample reads were assembled by using mothur v1.3213. Chimeric sequences were removed using the USEARCH software based on the UCHIME algorithm14. The microbial diversity was analyzed using the QIIME software15 with Python scripts. Operational Taxonomic Unit (OTUs) were picked using de novo OTU picking protocol with a 97% similarity threshold. Alpha diversity analysis included Shannon index, Chao1 and observed species. Jackknifed beta diversity included both unweighted and weighted Unifrac distances calculated with 10 times of subsampling and these distances were visualized by Principal Coordinate Analysis (PCoA)16. Taxonomy assignment of OTUs was performed by comparing sequences to the Greengenes database (gg_13_5_otus).
Mann-Whitney U test was used for significance test of alpha diversity. Two-sided Student's t-test was used for significance test of beta diversity difference between sample groups. Linear discriminant analysis coupled with effect size (LEfSe) was performed to identify the bacterial taxa differentially represented between groups at genus or higher taxonomy levels17. The functional profiles of microbial communities were predicted by using PICRUSt18. Bootstrap Mann-Whitney u-test with 1000 permutations was also used to identify gene pathways or OTUs with significantly different abundance between groups. The R packages “Phyloseq”, “biom”, “pheatmap” were used for data analysis and plotting19,20.
Results
Metadata and sequencing
Not surprisingly, rex rabbit body weights were significantly different between HW and LW groups (Figure S3) on both day 70 and day 90. A total of 40 fecal samples (10 HW hard feces, 10 HW soft feces, 10 LW hard feces and 10 LW soft feces) were collected and sent for sequencing. After OTU picking and chimera checking, a total of 2,078,821 reads were assigned to 60,783 non-singleton OTUs, which resulted in the classification of 474 taxa (genus level). Each sample has 6,904 OTUs and 51,970 sequences on average (Table S1).
Differences in bacterial communities between high and low weight rex rabbits
Three alpha diversity measures were calculated including Shannon's diversity index, observed species (observed OTUs) (Figure 1A) and Chao1 (estimated OTUs) (Figure S4). We found no significant difference in Shannon diversity between high weight (HW) and low weight (LW) samples (Figure 1A). For community richness comparison, low weight hard feces had significantly higher number of observed and estimated (Chao1) OTUs than high weight hard feces (p <0.01, Figure 1A and Figure S4). No significant differences in richness were observed between LW soft and HW soft feces.

Differences in bacterial community diversity, richness and structures between high and low weight rabbits.
(A): Community diversity and richness between high and low weight feces (both hard and soft feces). (B): Principal Coordinate Analysis (PCoA) of bacterial community structures of the gut microbiota of the four sample groups. Each symbol represents each gut microbiota. Bigger symbols represent high weight rabbits. Squares and circles represent hard and soft feces, respectively. (C): PCoA shows distinct bacterial communities between high and low weight hard feces. (D): No significant differences in bacterial communities were observed between high and low Weight soft feces. Sequences were normalized to the depth of 21,522 sequences with 10 times of subsampling to minimize the effect of sequencing depth. Asterisk shows significant differences between groups (** p <0.01, * p <0.05, Mann-Whitney U test).
The relationships between the community structures of the rex rabbit gut microbiota were examined by using the Principal Coordinate Analysis (PCoA) based on the unweighted and weighted Unifrac distance matrixes. On the PCoA plot, each symbol represents the gut microbiota of a rex rabbit (Figure 1B). Interestingly, the microbiotas of the LW hard feces were distinct from those of the HW hard feces (Figure 1B and C). No significant differences in community structure were observed between the HW and LW soft feces (Figure 1D). The relationships between community structures revealed by PCoA were further tested by comparing the between- and within-group unweighted Unifrac distances. Consistent with the PCoA plot, the between-group distances were significantly higher than the within-group distances (two-tailed Student's t-test, p <0.01) for the HW hard and LW hard pair, but not for the HW soft and the LW soft pair (Figure S5). These data suggests that the microbial community structures between HW and LW hard feces were significantly different whereas those between HW vs LW soft were not significantly different (Figure 1 and Figure S5).
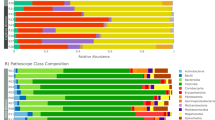
Figure 2 shows the community composition of the HW and LW rabbits in the two fecal types. In the stacked bar chart, each bar represents the average relative abundance of each bacterial taxon. The top 20 taxa with high relative abundance, which are in total composed of 93.4% of the reads, were illustrated.

Microbial composition of high and low weight rabbits in different fecal types.
Each bar represents the average relative abundance of each bacterial taxon within a group. The top 20 abundant taxa are shown.
To identify bacterial taxa that were significantly differentiated between groups, we performed LEfSe on 93 top taxa (average relative abundance> 0.0001). This threshold allowed us to keep as many taxa as possible for meaningful comparisons and to eliminate most rare taxa in the analysis. Figure 3 shows bacterial taxa differentially represented between high weight and low weight rabbits. In hard feces, 41 bacterial taxa were significantly more abundant in high weight rabbits (e.g. YS2, Bacteroidales, Lactococcus spp., Lactobacillus spp., Prevotella spp., Sutterella spp., Acinetobacter spp. p <0.05), while only 6 taxa were overrepresented in low weight rabbits (e.g. Anaeroplasma spp., Clostridiaceae, p <0.05) (Figure 3A). As to soft feces, only 3 differentially represented bacterial taxa were detected, with two and one taxa more abundant in high weight and low weight group (p <0.05), respectively (Figure 3B).

Bacterial taxa significantly differentiated between the high and low weight rabbits identified by linear discriminant analysis coupled with effect size (LEfSe) using the default parameters.
(A) and (B) show different taxa between the high weight and low weight rabbits in the hard and soft feces, respectively.
We next used a computational tool, PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)20, to explore the functional profiles of the rex rabbit gut microbiota. The 1123 closed-reference picked and differentially represented OTUs were normalized by 16S rRNA copy number and their metagenomic contributions were predicted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. In comparison between groups with different weight, 14 pathways (e.g. “Stilbenoid, diarylheptanoid and gingerol biosynthesis”, “Mineral absorption”, “Staphylococcus aureus infection”) were significantly more abundant in the hard feces of high weight rabbits, while other three infectious diseases related pathways (“Pathogenic Escherichia coli infection”, “Shigellosis”, “Bacterial invasion of epithelial cells”) were overrepresented in low weight rabbits (Figure 4, p<0.01) and no different pathways were found in their soft feces.

Predicted function of gut micorbiota between the high and low weight hard feces.
The gene copy numbers of samples within the same sample group were pooled. Values of each functional gene (row) were log2 transformed. The third level of KEGG pathway was shown in the heatmap. The significant test of the gene distribution between groups were performed using bootstrap Mann-Whitney u-test with cutoffs of p <0.01, FDR <0.1, Mean counts> 10.
Differences in bacterial communities between fecal types
When comparing alpha diversities between fecal types, no significant differences in diversity or richness were observed between hard and soft feces except a higher Chao1 in hard feces (LW) compared to soft feces (LW) (p <0.05, Figure 5A and Figure S3). Whereas PCoA plot based on unweighted Unifrac shows distinct bacterial community structures between hard and soft feces in high weight feces (Figure 5B), much larger and remarkable differences in community structures between fecal types were observed in low weight rabbits (Figure 5C).

Differences in bacterial community diversity, richness and structures between soft and hard feces.
(A): Community diversity and richness between soft and hard feces in high and low weight rabbits. (B): Principal Coordinate Analysis (PCoA) shows distinct bacterial community structures between the hard and soft feces of high weight rabbits. Each symbol represents each gut microbiota. Bigger symbols represent high weight rabbits. Squares and circles represent hard and soft feces, respectively. (C): PCoA shows much more striking differences in bacterial communities between the soft and hard feces in low weight rabbits. Sequences were normalized to the depth of 21,522 sequences with 10 times of subsampling to minimize the effect of sequencing depth.
We also performed LEfSe to detect bacterial taxa with significantly different abundance between fecal types (Figure 6). Eleven and forty four taxa were overrepresented in soft feces (e.g. Ruminococcaceae, Akkermansia spp., Blautia spp., Lactococcus spp., Barnesiellaceae, p <0.05) of high weight rabbits and low weight rabbits, respectively. Seven and eight taxa were identified to be more abundant in hard feces (e.g. S24-7, RF39, p <0.05) of high and low weight groups, respectively.

Bacterial taxa significantly differentiated between hard and soft feces identified by linear discriminant analysis coupled with effect size (LEfSe) using the default parameters.
(A) and (B) show different taxa between the hard and soft feces in the high weight and low weight rabbits, respectively.
Similarly, we used PICRUSt to explore the different metabolic potentials between fecal types. A total of 26 pathways (e.g. “Various types of N-glycan biosynthesis”) were more abundant in the soft feces and only 4 pathways were more abundant in hard feces (p <0.01). Interestingly, four “xenobiotics biodegradation” pathways (Xylene, Atrazine, Dioxin, Styrene) were enriched in soft feces (Figure 7, bootstrap Mann-Whiteney u-test, p <0.01).

Predicted function of gut micorbiota between the hard and soft feces in high weight (A) and low weight (B) rabbits.
The gene copy numbers of samples within the same sample group were pooled. Values of each functional gene (row) were log2 transformed. The third level of KEGG pathway was shown in the heatmap. The significant test of the gene distribution between groups were performed using bootstrap Mann-Whitney u-test with cutoffs of p <0.01, FDR <0.1, Mean counts> 10.
Discussion
Microbiota related to rabbit body weight
In this study we characterized the gut microbiota in rex rabbits with respect to their correlation with growth and feces types. We found several bacterial taxa such as YS2/Cyanobacteria, Bacteroides, Lactococcus spp., Lactobacillus spp., and Prevotella spp. were overrepresented in high weight hard feces (Figure 3A). Lactococcus spp. and Lactobacillus spp., members of which are well-known lactate producing probiotics, are wildly used to improve animal digestion efficiency. YS2 was previously recognized as a member of Cyanobacteria. Recent genomic study showed YS2 does not have phytosynthetic ability and classified this bacterium into the new class “Melainabacteria”. Metabolic analysis demonstrated YS2 has many special functions including obligate anaerobic fermentation, syntrophic H2-production, nitrogen fixation and synthesis of vitamin B and K21.
In human studies, the abundance of Bacteroides was reduced in obese children (e.g. Kazakh school children, preschool children of Estonia)22,23,24. However, our study suggests that members of Bacteroides were significantly enriched (p <0.05) in high weight rabbits. Therefore, not all the members of Bacteroides are negatively correlated with body weight. It is also important to note that the high weight rex rabbits are not obese. As small herbivores, rex rabbits contain much lower average fat contents than human. Therefore, members of Bacteroides might contribute to the healthy growth of rex rabbits.
Recently, Looft et al. (2014) studied the effects of antibiotic on swine gut microbiota and found that Carbadox pre-treatment prevented the increase of E. coli populations and promoted a large increase in relative abundance of Prevotella populations in these medicated pigs25. Consistently, our data also showed increased Prevotella and decreased Enterobacteriaceae (Figure 3A) in HW rabbits, mirroring the Carbadox mediated growth promotion effect26,27. Further investigation is warranted to confirm the effect of the differentially represented bacterial taxa in HW and LW groups on the growth of rex rabbits. Based on these studies, it is promising to then develop prebiotics and/or probiotics to promote the “growth positive” and inhibit the “growth-negative” bacterial taxa for the rex rabbits production.
With respect to metabolic pathways, the enrichment of “stilbenoid, diarylheptanoid and gingerol biosynthesis” and “Mineral absorption” pathways in the high weight rex rabbits group are remarkable. Stilbenoid diarylheptanoid and gingerol, known as plant source of phytoalexins were reported to have natural anti-inflammatory or anti-cancer functions28,29,30. Meanwhile, we also found an overrepresentation of “alpha-Linolenic acid metabolism” in hard feces of high weight rabbits (Figure 7A). Alpha-linolenic acid as one of the essential human fatty acids, is often used as nutritional supplement. It can be converted into Eicosapntemacnioc acid (EPA), Docosahexenoic acid (DHA) etc. in the body and lead to many health related functions like anti-inflammatory effects, or prevention of stroke and heart disease31,32,33. The enrichment of these health-related pathways might contribute to the higher weight of rex rabbits. Besides, the high abundance of infectious diseases pathways in low weight rabbits was construable due to its high Enterobacteriaceae content. However, none of the rabbits had symptoms associated with bacterial infection. Therefore, either these pathways were not expressed, or did not reach a level to cause the onset of symptoms.
Further experiments such as transcriptomics, metabolomics and fecal transplant are needed to verify the functions and roles that these high-weight enriched bacteria play in rabbit growth.
Specific gut bacteria in rabbit soft feces
Although similar microbial richness was observed between hard and soft feces, the community structures are significantly different between fecal types. Both in the HW and LW rabbit group, the soft fecal microbiota is strikingly separated from the hard fecal microbiota. Members of the Akkermansia spp., Blautia spp. and Oscillospira spp., are three of the several bacterial genera that were overrepresented in the soft fecal microbiota.
Akkermansia spp. has been detected in the intestines of many vertebrates such as human34, mouse35 and zebrafish36. Recent studies suggested Akkermansia spp. plays an important role in mucus degradation and production37. The enrichment of Akkermansia in rabbit soft feces is in keeping with the hypothesis that members of this bacterial taxon might contribute to the formation of the mucus-covered soft feces.
Blautia sp. has been proved to be one of the core microflora of many mammals. Although the relative abundance of Blautia spp. negatively correlated with many diseases such as Clostridium difficile infection38, type 1 diabetes39, acute hemorrhagic diarrhea40, cirrhosis41, some species of this genus such as B. faecis and B. stercoris can utilize carbohydrates as fermentable substrates and produce acetate and lactate as the major end products of glucose fermentation42,43. In addition, Godwin (2013) reported the B. coccoides as a reductive acetogen which are associated with carbon dioxide and hydrogen metabolism and results in reduced methane output of kangaroos44. Consistent with these studies, the enrichment of Blautia spp. in rabbit soft feces indicates members of this genus might play roles in the digestion of the diets in cecum.
Oscillospira spp. has been detected in the rumen of several herbivores such as cattle, sheep and reindeer45. Recently, members of this bacterium have been shown to be prevalent in macropod46 and humans47 as well. The overrepresentation of this genus in rabbit soft feces indicates that members of this genus might be involved in fermentation, as soft feces are the product of the caecum fermentation.
Several metabolic pathways were enriched in soft feces including bile secretion, mineral absorption and xenobiotics biodegradation. Bile secretion and mineral absorption belong to the digestive system pathway, although their relationship with gut bacteria is still unclear. We found four pathways related to xenobiotics biodegradation are enriched in soft feces, including atrazine, dioxin, styrene and xylene degradation. Atrazine is a broad-spectrum herbicide. Dioxin is also wildly contained in many pesticides. These compounds are considered toxic, mutagenic and possibly carcinogenic. The enrichment of these pathways in soft feces suggests that the gut bacteria in rabbit cecum may also have detoxification function.
The enrichment of “Various types of N-glycan biosynthesis” pathway in soft feces may be due to the presence of higher abundant Campylobacter spp. in soft feces (Figure 6B). This pathway, first discovered in Campylobacter jejuni48, directly related to N-linked protein glycosylation which influences multiple protein functions including sorting, targeting, localization, stability and quality control of protein synthesis. These functions were proved to enhance bacterial fitness by protecting bacterial proteins from cleavage by the gut proteases49.
It is worth noting that the accuracy of PICRUSt in predicting bacterial metabolic pathways depends much on the available reference bacterial genomes in the database. Although high agreement has been reached between PICRUSt predictions and human metagenome data across all body sites, the relevance of application of PICRUSt to predict bacterial activities in rabbits gut needs further validation. The average Nearest Sequenced Taxon Index (NSTI, 0.195 ± 0.05 s.d.) of our samples is comparable to those of other mammals (0.14 ± 0.06) and soils (0.17 ± 0.02). Despite the accurate metagenome predictions for soil samples and a subset of mammals (NSTI<0.05), the prediction accuracy of PICRUSt in rabbits needs further validation. Furthermore, a large portion of the OTUs were not matched to the database, thus their functions were not imputed. Of note, some human related pathways were identified probably due to poor annotation in the KEGG database and/or homologous pathways between bacteria and humans. These pathways were not discussed in this study. Nevertheless, despite these potential biases, PICRUSt provide important insight into bacterial community functions in rabbit gut. Other omics approaches (e.g. transcriptomics and metabolomics) are desired to confirm these discoveries and improve our understanding of the bacterial functions in rabbit guts.
In summary, we characterized the gut microbiota in soft and hard feces of rex rabbits with different weight. We identified bacterial taxa and metabolomic pathways that were overrepresented in different fecal groups. These features serve as immediate targets for future studies to test their roles in the growth and caecotrophic behavior of rex rabbits.
References
Hirakawa & Hirofumi Coprophagy in leporids and other mammalian herbivores. Mammal Rev 31, 61–80 (2001).
Chivers, D. J. & Langer, P. The digestive system in mammals: food, form and function, (Cambridge University Press, 1994).
Blas, C. D. & Wiseman, J. The nutrition of the rabbit, (CABI Publishing, 1998).
Kenagy, G. & Hoyt, D. F. Reingestion of feces in rodents and its daily rhythmicity. Oecologia 44, 403–409 (1979).
Sukemori, S., Ikeda, S., Kurihara, Y. & Ito, S. Amino acid, mineral and vitamin levels in hydrous faeces obtained from coprophagy-prevented rats. J Anim Physiol An N 87, 213–220 (2003).
Michelland, R. J. et al. Molecular analysis of the bacterial community in digestive tract of rabbit. Anaerobe 16, 61–5 (2010).
Abecia, L. et al. Molecular profiling of bacterial species in the rabbit caecum. FEMS microbiology letters 244, 111–115 (2005).
Cho, I. et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626 (2012).
Liou, A. P. et al. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Transl Med 5, 178ra41–178ra41 (2013).
Chung, H. et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593 (2012).
Zhang, C. et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat Commun 4, 2163 (2013).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624 (2012).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75, 7537–41 (2009).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–200 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–6 (2010).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microb 71, 8228–35 (2005).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60 (2011).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–21 (2013).
McMurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS One 8, e61217; 10.1371/journal.pone.0061217 (2013).
McDonald, D. et al. The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. GigaScience 1, 7 (2012).
Di Rienzi, S. C. et al. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife 2, e01102 (2013).
Xu, P., Li, M., Zhang, J. & Zhang, T. Correlation of intestinal microbiota with overweight and obesity in Kazakh school children. BMC Microbiol 12, 283 (2012).
Sepp, E., Loivukene, K., Julge, K., Voor, T. & Mikelsaar, M. The association of gut microbiota with body weight and body mass index in preschool children of Estonia. Microb Ecol Health D 24, 19231 (2013).
DiBaise, J. K. et al. Gut microbiota and its possible relationship with obesity. in Mayo Clin Proc Vol. 83 460–469 (Elsevier, 2008).
Looft, T., Allen, H. K., Casey, T. A., Alt, D. P. & Stanton, T. B. Carbadox has both temporary and lasting effects on the swine gut microbiota. Antimicrob Agents Ch 5, 276 (2014).
Laine, T. et al. The effect of antimicrobial growth promoter withdrawal on the health of weaned pigs in Finland. Prev Vet Med 66, 163–174 (2004).
Che, T. et al. Effect of dietary acids on growth performance of nursery pigs: A cooperative study. J Anim Sci 90, 4408–4413 (2012).
Morita, H., Koyama, K., Sugimoto, Y. & Kobayashi, J. I. Antimitotic activity and reversal of breast cancer resistance protein-mediated drug resistance by stilbenoids from Bletilla striata. Bioorg Med Chem Lett 15, 1051–1054 (2005).
Yadav, P. N., Liu, Z. & Rafi, M. M. A diarylheptanoid from lesser galangal (Alpinia officinarum) inhibits proinflammatory mediators via inhibition of mitogen-activated protein kinase, p44/42 and transcription factor nuclear factor-κB. J Pharmacol Exp Ther 305, 925–931 (2003).
Park, K.-K., Chun, K.-S., Lee, J.-M., Lee, S. S. & Surh, Y.-J. Inhibitory effects of [6]-gingerol, a major pungent principle of ginger, on phorbol ester-induced inflammation, epidermal ornithine decarboxylase activity and skin tumor promotion in ICR mice. Cancer Lett 129, 139–144 (1998).
Erdinest, N., Shmueli, O., Grossman, Y., Ovadia, H. & Solomon, A. Anti-inflammatory effects of alpha linolenic acid on human corneal epithelial cells. Invest Ophth Vis Sci 53, 4396–4406 (2012).
Nguemeni, C., Gouix, E., Bourourou, M., Heurteaux, C. & Blondeau, N. Alpha-linolenic acid: A promising nutraceutical for the prevention of stroke. PharmaNutrition 1, 1–8 (2013).
Mozaffarian, D. Does alpha-linolenic acid intake reduce the risk of coronary heart disease? A review of the evidence. Altern Ther Health M 11, 24–30 (2004).
Derrien, M., Vaughan, E. E., Plugge, C. M. & de Vos, W. M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Micr 54, 1469–76 (2004).
Presley, L. L., Wei, B., Braun, J. & Borneman, J. Bacteria associated with immunoregulatory cells in mice. Appl Environ Microb 76, 936–41 (2010).
Roeselers, G. et al. Evidence for a core gut microbiota in the zebrafish. ISME J 5, 1595–608 (2011).
Belzer, C. & de Vos, W. M. Microbes inside--from diversity to function: the case of Akkermansia. ISME J 6, 1449–58 (2012).
Shankar, V. et al. Species and genus level resolution analysis of gut microbiota in Clostridium difficile patients following fecal microbiota transplantation. Microbiome 2, 13 (2014).
Murri, M. et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med 11, 46 (2013).
Suchodolski, J. S. et al. The fecal microbiome in dogs with acute diarrhea and idiopathic inflammatory bowel disease. PLOS One 7, e51907 (2012).
Kakiyama, G. et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 58, 949–955 (2013).
Park, S.-K., Kim, M.-S. & Bae, J.-W. Blautia faecis sp. nov., isolated from human faeces. Int J Syst Evol Micr 63, 599–603 (2013).
Park, S.-K., Kim, M.-S., Roh, S. W. & Bae, J.-W. Blautia stercoris sp. nov., isolated from human faeces. Int J Syst Evol Micr 62, 776–779 (2012).
Godwin, S. et al. Investigation of the microbial metabolism of carbon dioxide and hydrogen in the kangaroo foregut by stable isotope probing. ISME J 8, 1855–1865 (2014).
Mackie, R. I. et al. Ecology of Uncultivated Oscillospira Species in the Rumen of Cattle, Sheep and Reindeer as Assessed by Microscopy and Molecular Approaches. Appl Environ Microb 69, 6808–6815 (2003).
Gulino, L. M. et al. Shedding light on the microbial community of the macropod foregut using 454-amplicon pyrosequencing. PLOS One 8, e61463 (2013).
Tims, S. et al. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J 7, 707–17 (2013).
Kelly, J. et al. Biosynthesis of the N-linked glycan in Campylobacter jejuni and addition onto protein through block transfer. J Bacteriol 188, 2427–2434 (2006).
Alemka, A., Nothaft, H., Zheng, J. & Szymanski, C. M. N-glycosylation of campylobacter jejuni surface proteins promotes bacterial fitness. Infect Immun 81, 1674–1682 (2013).
Acknowledgements
This work was supported by the“1000-Talent Program” in Sichuan and Science Foundation for Youths of Sichuan Province (2013JQ0014) and The National Natural Science Foundation of China (31471997) to Y.L. and the Innovative Research Team in University of Sichuan Bureau of Education. B. Z. was supported by the applied fundamental research program of Sichuan province (2014jy0135). P.W. was supported by the earmarked fund for China Agriculture Research System (CARS-44-A-4) and the Project of Provincial Twelfth 5 Years' Rabbit Breeding of Sichuan Province (2011NZ0099-4).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: B.Z., P.W., S.H., J.Z. and Y.L. Performed the experiments: B.Z., P.W. and S.H. Contributed reagents/materials/analysis tools: B.W., W.J., Z.Y., D.D., W.G., X.F., F.K., X.S. and M.Y. Wrote the paper: B.Z., J.Z. and Y.L.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zeng, B., Han, S., Wang, P. et al. The bacterial communities associated with fecal types and body weight of rex rabbits. Sci Rep 5, 9342 (2015). https://doi.org/10.1038/srep09342
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09342
This article is cited by
-
Arula-7 powder improves diarrhea and intestinal epithelial tight junction function associated with its regulation of intestinal flora in calves infected with pathogenic Escherichia coli O1
Microbiome (2023)
-
Rabbit microbiota across the whole body revealed by 16S rRNA gene amplicon sequencing
BMC Microbiology (2021)
-
Individual fate and gut microbiome composition in the European wild rabbit (Oryctolagus cuniculus)
Scientific Reports (2021)
-
Dynamic distribution of gut microbiota in meat rabbits at different growth stages and relationship with average daily gain (ADG)
BMC Microbiology (2020)
-
Comparison Between the Gut Microbiota in Different Gastrointestinal Segments of Large-Tailed Han and Small-Tailed Han Sheep Breeds with High-Throughput Sequencing
Indian Journal of Microbiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
