
Abstract
This study aimed to investigate the possible therapeutic effects and active components of Lycium barbarum polysaccharides (LBP) on a high fat diet-induced NASH rat model. We induced NASH in a rat model by voluntary oral feeding with a high-fat diet ad libitum for 8 weeks. After 8 weeks, 1 mg/kg LBP was orally administered for another 4 weeks with a high-fat diet. When compared with NASH rats treated for 12 weeks, therapeutic LBP treatment for 4 weeks during 12 weeks of NASH induction showed ameliorative effects on: (1) increased body and wet liver weights; (2) insulin resistance and glucose metabolic dysfunction; (3) elevated level of serum aminotransferases; (4) fat accumulation in the liver and increased serum free fatty acid (FFA) level; (5) hepatic fibrosis; (6) hepatic oxidative stress; (7) hepatic inflammatory response; and (8) hepatic apoptosis. These improvements were partially through the modulation of transcription factor NF-κB, MAPK pathways and the autophagic process. In a palmitate acid-induced rat hepatocyte steatosis cell–based model, we also demonstrated that l-arabinose and β-carotene partially accounted for the beneficial effects of LBP on the hepatocytes. In conclusion, LBP possesses a variety of hepato-protective properties which make it a potent supplementary therapeutic agent against NASH in future clinical trials.
Similar content being viewed by others

Introduction
Non-alcoholic fatty liver disease (NAFLD) is one of the most common liver diseases in the world, particularly in affluent areas. It is not only linked to other metabolic disorders (e.g. type 2 diabetes), but it also contributes to the late-stage liver diseases including cirrhosis and hepatocellular carcinoma (HCC)1. When inflammatory and fibrotic responses are activated in the liver, NAFLD progresses to non-alcoholic steatohepatitis (NASH) which may also induce severe cellular oxidative stress, necrosis and apoptosis2. To date, the detailed pathological mechanisms for the development of NASH remain poorly understood. Recently, a “multiple-parallel-hit” theory was proposed in which dysregulated lipid metabolism and insulin resistance are the first hit to the liver, followed by a “second-hit” or “multi-hits” such as oxidative stress, inflammation, apoptosis and fibrosis3. Currently, there are two major therapeutic strategies for NAFLD/NASH: (1) lifestyle interventions (including weight reduction, dietary modification and physical exercise) and (2) pharmaceutical therapy. However, a successful management is based on appropriate combination of these therapies according to the personal variation of specific patient4.
During the past decades, applying natural products to treat NASH received wide attention since some of them exhibited effective outcomes with controlled side effects, including coffee5, garlic6,7, green tea8, milk thistle9 and resveratrol10. Polysaccharide portion from wolfberry (Lycium barbarum polysaccharides, LBP) is a new medicinal herbal derivative, which is reported to possess positive effects in nourishing the liver and maintaining the health of the eyes11. It also plays a protective role in hepatoma cancer cell line12 and the neuron system13. Recently, we have reported that LBP protected rodent's liver from carbon tetrachloride-induced acute liver injury14 and high-fat diet-induced NASH15. Although we demonstrated that co-administration with LBP evidently attenuated hepatic steatosis, fibrosis, oxidative stress, inflammation and apoptosis in NASH rats, we still do not know (1) whether LBP could play a therapeutic role against the development of NASH; and (2) what are the effective specific monomers that account for the hepatic beneficial effects of LBP15? In the current study, we mainly focused on the therapeutic effects and mechanisms of LBP on an already established NASH rat model. In addition, we also applied an in vitro steatosis cell-based model in normal rat hepatocyte line BRL-3A and investigated the possible protective effects of two LBP components, such as l-arabinose and β-carotene.
Methods
Reagents and antibodies
All cell culture consumables and reagents were bought from either Corning Incorporated (Corning, NY) or Gibco (Carlsbad, CA). Antibodies against phosphorylated SMAD2 at Ser467, total SMAD2, total SMAD4 and β-actin were bought from Santa Cruz Biotechnology (Santa Cruz, CA). Phosphorylated SMAD4 at Thr277 antibody was purchased from Abnova (Taipei, Taiwan). Resistin antibody was ordered from Abcam (Cambridge, UK). Antibodies of nitrotyrosine, phosphorylated IRS-1 at Ser307, total IRS-1, phosphorylated GSK3α at Ser21, total GSK3α, phosphorylated p38 MAPK at Thr180/Tyr182, total p38 MAPK, phosphorylated JNK at Thr183/Tyr185, total JNK, phosphorylated ERK1/2 at Thr202/Tyr204, total ERK1/2, IκBα, cleaved caspase-3, cytochrome c, p62, beclin-1, Atg5, phosphorylated mTOR at Ser2448 and total mTOR were purchased from Cell Signaling (Beverly, MA). Synaptophysin antibody was bought from DAKO (Glostrup, Denmark).
Animal experiments
Healthy adult female Sprague-Dawley (SD) rats with a body weight ranging from 180–200 g were obtained from the Laboratory Animal Unit (LAU), The University of Hong Kong. Rats were maintained and cared for in compliance with the requirements of The University of Hong Kong and the National Institute of Health guidelines. The entire experimental procedures were conducted according to the protocols approved by the Committee of Animal Use for Research and Teaching at The University of Hong Kong. The Laboratory Animal Unit of The University of Hong Kong is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International).
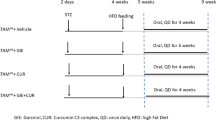
The animals were divided into six groups (n = 6 per group) namely: (1) control group (rats were fed with regular rat diet and tap water for 12 weeks); (2) vehicle-LBP group (1 mg/kg LBP in 1 × PBS; oral gavage feeding; one time per day during the entire experimental duration); (3) vehicle-therapeutic LBP group (1 mg/kg LBP in 1 × PBS; oral gavage feeding; one time per day from the 9th week to the 12th week); (4) NASH group (rats were fed with a high-fat diet for 12 weeks); (5) NASH + LBP group (high-fat diet feeding with 1 mg/kg LBP in 1 × PBS; oral gavage feeding; one time per day during the entire experimental duration); and (6) NASH + therapeutic LBP group (high-fat diet feeding with 1 mg/kg LBP in 1 × PBS; oral gavage feeding; one time per day from the 9th week to the 12th week). The entire experimental duration was 12 weeks. Open liver biopsy was performed on the 8th week under intraperitoneal injection of anesthesia (Ketamine and Xylazine). The development of NASH in rats, including the recipe and preparation protocols of diet, was performed based on our previously described voluntary oral feeding NAFLD animal model15,16. The diet consists of 9.3 g AIN-93MX (Dyets incorporation, Bethlehem, PA), 2.6 g AIN-93VX (Dyets), 0.5 g choline bitatrate (Dyets), 1.1 g DL-methione (Bio-serv, Frenchtown, NJ), 57.5 g lactalbulmine hydrolysate (Bio-serv), 117.5 g dextrose (Dyets), 36.6 ml fish oil (Sigma) and 4.5 g suspending agent K (Bio-serv) in per 1,000 ml volume. Regular chow for rat (PicoLab® Rodent Diet 20) was purchased from LabDiet (LabDiet, Brentwood, MO). The calories of regular chow were provided by 25% from protein, 13% from fat and 62% from carbohydrates, while the calories of high-fat diet were provided by 35% from protein, 30% from fat and 35% from carbohydrates. The selection of LBP dose was based on our previous studies on both acute and chronic liver models14,15. After twelve weeks, rats were killed by an overdose of anesthesia (150 mg/kg pentobarbital, intraperitoneal injection) according to the protocols approved by the Committee of Animal Use for Research and Teaching at The University of Hong Kong after a 12-hr fasting. Blood and liver samples were collected for further analyses.
Processing of blood and tissue samples
Serum was collected by centrifugation from whole blood sample at 1,000 × g for 10 min at 4°C and stored at −80°C. Liver tissue samples were fixed in 10% phosphate-buffered formalin, processed for histology and embedded in paraffin blocks. Five-micrometer tissue sections were cut and stained with hematoxylin and eosin (H&E), Sirius red and synaptophysin (DAKO, Denmark) for histological analysis using a LEICA Qwin Image Analyser (Leica Microsystems Ltd., Milton Keynes, UK). The NAFLD activity score (NAS) of each group was calculated as previously described17.
Serum ALT and AST assay
To evaluate the hepatic injury at the enzymatic level, serum ALT and AST levels were measured by using ALT (SGPT) and AST (SGOT) reagent sets (Teco diagnostics, Anaheim, CA) according to manufacturer's instructions.
Free fatty acids (FFA) assay
To study the effect of LBP on lipid metabolism, serum FFA level of each rat was measured by using a Cayman free fatty acids assay kit (Cayman chemical, Ann Arbor, MI) and the final results were expressed as μM in the serum.
Measurement of MDA level
Levels of the end-product of lipid peroxidation (malondialdehyde, MDA) in all liver tissue samples were determined by using a Bioxytech LPO-586™ kit (Oxis Research, Portland, OR). The reaction product was measured spectrophotometrically at 586 nm. Standard curves were constructed using 1,1,3,3-tetraethoxypropane as a standard. The MDA levels were normalized with corresponding protein amounts determined by a Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA) and expressed as percentage against the control level.
Insulin tolerance test (ITT) and glucose tolerance test (GTT)
Measurements of ITT and GTT were performed as previously described18 with modifications in accordance to the Vanderbilt recommendations for evaluating glucose homeostasis19. Briefly, after 12-week NAFLD induction, rats were placed in clean cages under starvation state but with water supply for 6 hrs. Then rats subjected to ITT or GTT assays were intraperitoneally (i.p.) injected with recombinant insulin (a fixed bolus of 0.17 IU, R&D systems, Minneapolis, MN) or D-glucose (a fixed bolus of 350 mg, Sigma-Aldrich, St. Louis, MO), respectively. We used a fixed bolus of insulin or glucose because lean body mass (e.g. muscle, liver and brain) was the main site of glucose disposal, even though obesity was primarily caused by an increase in fat mass. Administering glucose on a per body weight basis would increase the glucose dose disproportionately in relation to the major glucose-utilizing tissue19. 0, 20, 40, 60, 80 and 100 min after insulin/glucose injection, tail vein blood sample from each rat was tested for glucose level directly by using ACCU-CHEK blood glucose monitoring system (Roche Diagnostic, Basel, Switzerland). Serum insulin levels were quantified by using the commercial ELISA kits from Antibody and Immunoassay Services of The University of Hong Kong (Hong Kong, China). Basal plasma glucose concentrations were also recorded to normalize the results of GTT19.
ELISA assay
ELISA measurements of TNF-α, IL-1β and MCP1 were performed by using corresponding ELISA development kits from PeproTech (PeproTech Inc., Rocky Hill, NJ) according to user instructions. COX-2 ELISA assay was conducted with kit from EIAab (Wuhan EIAab Science, Wuhan, China). TGF-β1 and α-SMA ELISA kits were from eBioscience (San Diego, CA) and MyBioSource (San Diego, CA), respectively.
Western blot
Western blot analyses of liver tissue extracts were performed as described6. The ratio of the optical density of the protein product to the internal control (β-actin) was obtained and was expressed as ratio or percentage of the control value in the Figures.
NF-κB activity
The activity of transcription factor NF-κB p65 was measured with a NF-κB/p65 ActivELISA kit from IMGENEX Corporation (San Diego, CA) according to user's instructions.
Cell culture and treatments
The rat hepatocyte BRL-3A cell line was supplied by the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI-1640 with 10% (v/v) FBS at 37°C and 5% CO2 using a cell incubator. When cells reached a confluence of 60–70%, they were divided into 8 groups (n = 5) including: (1) control group (with PBS which was the dissolvent for LBP); (2) steatosis group [treated with palmitate acid (PA)]; (3) vehicle-LBP group (treated with LBP only); (4) steatosis + LBP group (treated with PA and LBP); (5) vehicle-arabinose group (treated with 3 mM l-arabinose only); (6) steatosis + arabinose group (treated with PA and 3 mM l-arabinose); (7) vehicle-carotene group (treated with 2 μM β-carotene only); and (8) steatosis + carotene group (treated with PA and 2 μM β-carotene). The treatment duration was 24 hrs. The lipid accumulation in each group was revealed by Oil Red O staining. The dose selection of l-arabinose20 and β-carotene21 was based on previous in vitro reports.
MTT assay
Cell viability was evaluated by the conversion of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich) to a purple color product by cellular mitochondria. After drug treatment, cells from each group were washed by sterile PBS 3 times and then incubated with 5 mg/ml MTT for 3 hrs and subsequently dissolved in dimethyl sulfoxide (DMSO). The absorbance of MTT was measured at 570 nm.
Glucose production assay
After treatment, cells were washed by sterile PBS for 3 times to remove glucose and then incubated for 16 hrs in glucose production medium (glucose- and phenol red-free DMEM containing gluconeogenic substrates, 20 mM sodium lactate and 2 mM sodium pyruvate) in the presence of 1 nM insulin (R&D systems) during the last 4 hrs. The concentration of glucose was measured using a commercial kit from Sigma. Glucose concentration was normalized with corresponding cellular protein concentration.
Quantification of apoptotic cells
After drug treatment, Hoechst 33342 (5 μg/ml) and propidium iodide (5 μg/ml) were added to each well to stain live cells. The results were expressed as the percentage of apoptosis (PA): PA = apoptotic cell number/total cell number × 100%22.
Activities of caspase- 3/7
Activities of caspases-3 and -7 from both liver tissue lysates and cell lysates were measured using Caspase-Glo 3/7 Assay Systems (Promega, Madison, WI) according to the user's manual. The luminescence was read in a Glomax luminometer (Promega) and expressed as fold of change in caspase 3/7 activity from the control.
RNA extraction and quantitative PCR
Total RNA of each rat was extracted from the liver sample or cell lysate by an using illustra™ RNAspin mini kit (GE healthcare, UK). The preparation of the first-strand cDNA was conducted following the instruction of the SuperScript™ First-Strand Synthesis System (Invitrogen, Calsbad, CA). The mRNA expression levels of target genes were measured by a Takara SYBR premix Taq quantitative PCR system (Takara Bio Inc, Shiga, Japan) using a MyiQ2 real-time PCR machine (Bio-Rad). The primer sequences and annealing temperatures used in these real-time PCR reactions are listed in Suppl. Table S1. Parallel amplification of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control. Relative quantification was done by using the 2−ΔΔCt method. The relative expression of the specific gene to the internal control was obtained and then expressed as a percentage of the control value in the Figures. All real-time PCR procedures including the design of primers, validation of PCR environment and quantification methods were performed according the MIQE guideline23.
Statistical analysis
Data from each group were expressed as means ± SEMs. The trapezoidal rule was used to determine the area under the curve (AUC). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. A p < 0.05 was considered to be statistically significant (Prism 5.0, Graphpad software, Inc., San Diego, CA, USA).
Results
Confirmation of NASH induced by an 8-week high-fat diet feeding
To make sure the successful establishment of NASH lesions in the rat liver, liver biopsy of high-fat diet fed rats was performed at the end the 8th week, before the start of the “therapeutic” administration of LBP. It is shown that high-fat diet induced evident hepatic histopathological changes in the liver, such as lipid accumulation, foci of inflammatory cells and necrosis (Supplementary Figures 1A and 1B). The NAS score of the liver from the high-fat diet fed rats was significantly higher than those of the normal rats fed with a regular animal chow fed (Supplementary Figure 1C).
LBP treatments ameliorated rat obesity and insulin resistance
Eight-week consumption of high-fat diet induced significant increase in body weight of the rats when compared with the control rats fed with a regular rat diet (365.0 ± 9.8 g versus 326.3 ± 11.6 g at 8 weeks, p < 0.05). Administration of 1 mg/kg LBP significantly reduced the body weight of NASH rats from the 3rd week to the 12th week (340.2 ± 13.4 g [LBP-treated] versus 401.7 ± 10.7 g [NASH-treated] at the 12th week, p < 0.05). For the therapeutic treatment of LBP, which started from the 9th to 12th week, also significantly reduced the body weight of high-fat diet fed rats (352.1 ± 14.0 g [LBP-treated] versus 401.7 ± 10.7 g [NASH-treated] at the 12th week, p < 0.05) (Figure 1A). Administration of LBP in both LBP-treated groups (NASH-LBP 12 weeks and NASH-LBP 9-12week) did not influence the body weight when compared with the control rats (data not shown). Results of wet liver weight further confirmed the anti-obesity effect of LBP. The wet liver weight of NASH rats was significantly higher than that of the control rats (13.4 ± 0.1 g versus 11.5 ± 0.3 g, p < 0.05). Both LBP-treated groups reduced the liver weight of NASH rats comparable to control level (Figure 1B). Interestingly, for most weeks, administration of LBP did not influence the appetite of NASH rats. There was no significant difference in the daily food intake volume between LBP-treated NASH rats and NASH rats without LBP except for weeks 3, 4 and 11 (Figure 1C).

LBP therapeutically improved NASH-induced obesity, insulin resistance and glucose metabolism dysfunction.
(a) The body weight of each rat was recorded every week throughout the 12-week high-fat diet induction of NASH. (b) After the induction, the wet liver weight was weighed for each rat. (c) The food intake of each rat (in ml/d) was recorded everyday throughout the 12-week high-fat diet induction of NASH. Each dot represents the average food intake of a week. (d) Representative Western blot results of resistin, phosphorylated IRS-1, total IRS-1, phosphorylated GSK3α and total GSK3α in all group of rats from 3 repeated Western blot experiments. (e) Blood glucose level was assessed after an intraperitoneal (i.p.) injection with recombinant insulin (a fixed bolus of 0.17 IU) and the serum glucose level was recorded 0, 20, 40, 60, 80 and 100 min later after the 12-week induction of NASH. (f) Area under the curve for glucose (AUCglucose) was calculated using the trapezoidal rule. (g) Blood glucose level of each rat was recorded 0, 20, 40, 60, 80 and 100 min after an i.p. injection of D-glucose (a fixed bolus of 350 mg) after the 12-week induction of NASH. Data were normalized with basal glucose levels. (h) AUCglucose was calculated using the trapezoidal rule. Data from each group were expressed as means ± SEMs (n = 6). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. A p < 0.05 was considered to be statistically significant. “*” represents significant change against control and “#” represents significant change against either LBP co-treatment group or LBP after-treatment group. Different letters (e.g. a and b) mean a statistical significant change between each other.
In NASH rats, injection of recombinant insulin caused impaired insulin metabolism/clearance when compared with the control and vehicle-LBP rats (Supplementary Figure 2B). Treatments with LBP significantly improved the insulin clearance impairment induced by NASH (Supplementary Figure 2B). In addition, from the results of blood glucose and its AUC areas, LBP treatments effectively improved insulin resistance (Figure 1E and 1F). Similarly, after normalization with basal glucose level, we found that dysregulated glucose metabolism in NASH rats was also evidently corrected by LBP administration in both treated groups (Figure 1G and 1H, Supplementary Figure 2A). To further investigate the pathways involved in the improvement of insulin and glucose metabolism, we measured the change of resistin and IRS-1/GSK3α pathways in all groups. It was found that high-fat diet significantly increased the protein level of resistin and the phosphorylation level of GSK3α. It also decreased the phosphorylation of IRS-1. LBP administration, either co-treatment or delayed-treatment, counter-acted the effects of high-fat diet on these pathways without influencing the total protein level of IRS-1 and GSK3α (Figure 1D).
LBP treatments improved hepatic injury
Hepatic H&E staining results showed that 12-week administration of high-fat diet caused typical NASH pathological phenotypes of the rat's liver, including a large number of lipid droplet accumulation, infiltration of inflammatory cells and necrosis of hepatocytes (Figure 2A). All of the time points in LBP treatment (from week 1 to week 12) significantly improved the hepatic histopathology after 12 weeks and exhibited cellular architecture comparable to the control. Interestingly, therapeutic LBP treatment from week 9 to week 12 also dramatically ameliorated the severity of lipid deposition and reduced inflammatory foci and cellular necrosis in the liver. There was no statistical difference between the NAS score of two groups of LBP treatments (NASH-LBP 12 weeks and NASH-LBP 9-12week; Figure 2C). Sirius red staining found that the collagen formation in the liver of NASH rats was significantly more than those of the control rats. Both treatment groups of LBP effectively reduced the collagen formation, leading to attenuation of hepatic fibrosis (Figures 2B and 2D). Consistently, NASH rats exhibited the highest level of serum ALT level, which was significantly reduced by both groups of LBP treatment (Figure 2E). For the AST level, there was no significant variation among all groups of rats (Figure 2F).

LBP therapeutically ameliorated hepatic injury and fibrosis.
(a) Typical H&E staining results of each group of rat. Yellow arrows indicate hepatic fat accumulation and red arrow indicates typical inflammatory infiltration in the liver sections. Magnification: ×200. Bar: 20 μm. (b) Typical Sirius red staining results of each group of rat. Magnification: ×200. Bar: 20 μm. (c) NAFLD activity scores (NAS) of each group of rat after NASH induction and LBP treatments. (d) Collagen staining quantification of each group of rat after NASH induction and LBP treatments, as shown by the Sirius red staining. (e,f) Serum ALT and AST levels in each group. Data from each group were expressed as means ± SEMs (n = 6). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. Different letters (e.g. a and b) mean a statistical significant change (p < 0.05) between each other. TLBP: therapeutic LBP treatment from the 9th week to the 12th week with NASH induction.
LBP treatments alleviated hepatic fibrosis
To further investigate the molecular mechanisms underlying the reduction of hepatic fibrosis by LBP, the expressional change of the TGF-SMAD pathway was firstly assessed. The protein expression of both TGF-β1 and α-SMA was up-regulated by the induction of NASH (Figures 3A and 3B). Western blot assay also showed that the phosphorylation level of both SMAD2 and SMAD4 was increased in the NASH group (Figure 3C). Treatments with LBP, either with NASH-LBP 12 weeks or therapeutically (NASH-LBP 9–12 weeks), significantly reduced the phosphorylated level of these proteins without influencing their basal levels. In addition, since the formation of hepatic fibrosis is closely associated with the activation of hepatic stellate cells (HSCs), we then quantified the number of activated HSCs in each group by staining synaptophysin, a novel marker for activated HSCs in the liver24. The number of synaptophysin-positive in the NASH group was significantly higher than other five groups, predominantly around the centrolobular vein areas (Figure 3D). Both groups of LBP treatment (NASH-LBP 12 weeks and NASH-LBP 9-12 weeks) reduced the number of activated HSCs comparable to the control level (Figure 3E). LBP showed no obvious effect on the fibrotic change of healthy rats except a small reduction of the α-SMA protein level (Figure 3B).

Molecular pathways accounted for the anti-fibrotic effects of LBP.
(a,b) Expressional changes of hepatic TGF-β1 and α-SMA in each group of rat using ELISA kits. (c) Typical Western blot results of the expressional changes of SMAD2/4 in each group of rat from 3 repeated Western blot experiments. Full-length blots are presented in Supplementary Fig. 4. (d) Representative synaptophysin staining result in each group. Synaptophysin is a marker for activated hepatic stellate cells. Arrows indicate typical positive staining. Magnification: ×200. Bar: 20 μm. (e) Quantitative results for the synaptophysin staining. Data from each group were expressed as means ± SEMs (n = 6). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. Different letters (e.g. a and b) mean a statistical significant change (p < 0.05) between each other. TLBP: therapeutic LBP treatment from the 9th week to the 12th week with NASH induction.
Hepatic lipid accumulation and oxidative stress were attenuated by LBP
To explore the underlying mechanisms for the attenuated hepatic lipid accumulation by both groups of LBP treatment (NASH-LBP 12 weeks and NASH-LBP 9–12 weeks), expressional changes of lipogenic genes (SREBP-1c and PPARγ2) and lipolytic genes (ATGL and adiponectin) were measured. Induction of NASH significantly increased the hepatic mRNA expression of SREBP-1c and PPARγ2, while reduced the mRNA expression of ATGL and adiponectin, leading to a net increase of the serum FFA level (Figures 4A–4E). In line with the attenuated steatosis in the liver, the influence of NASH on lipid metabolic genes was abolished in both groups of LBP treatment (Figures 4A–4D). Thus, the serum level of FFA was regulated to the healthy rat level (Figure 4E). LBP treatments did not affect the rats without NASH development (Figures 4A–4E).

LBP therapeutically re-balanced lipid metabolism and attenuated hepatic oxidative stress.
(a,b) Expressional changes of lipogenic markers SREBP-1c and PPARγ2 in the liver of each rat, measured by quantitative PCR. (c,d) Expressional changes of lipolytic markers ATGL and adiponectin in the liver of each rat, measured by quantitative PCR. (e) Serum free fatty acid (FFA) level measured by a commercial kit in all rats after 12-week experiment. (f,g) The mRNA expressional changes of hepatic antioxidant enzymes catalase (CAT) and glutathione peroxidase (GPx) from each rat. (h) Hepatic malondialdehyde (MDA) level in each rat after 12-week experiment. (i) Representative Western blot results of nitrotyrosine formation in the liver of each rat from 3 repeated Western blot experiments. Full-length blots are presented in Supplementary Fig. 4. Data from each group were expressed as means ± SEMs (n = 5). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. Different letters (e.g. a and b) mean a statistical significant change (p < 0.05) between each other. C: control, N: NASH, L: vehicle-LBP, NL: NASH + LBP treatment throughout the experiment, TL: vehicle-LBP treatment from the 9th week to the 12th week with NASH induction, NTL: NASH + LBP treatment from the 9th week to the 12th week with NASH induction.
Lipid peroxidation and induced oxidative stress are common consequences of steatosis and insulin resistance during the progression of NASH25. To test the possible antioxidant property of LBP, the mRNA levels of endogenous antioxidant enzymes CAT and GPx were assessed firstly. In NASH rats, the hepatic level of these enzymes was significantly down-regulated, exhibiting an impaired defense against hepatic oxidative stress (Figures 4F and 4G). Administration of LBP, either along with or after the establishment of the NASH induction, effectively restored the levels of both antioxidant enzymes. Then we measured the products of lipid peroxidation (hepatic MDA content) and nitric oxide pathway (hepatic nitrotyrosine formation) in all groups of rats. NASH rats showed elevated content of both MDA and nitrotyrosine, indicating an increased status of hepatic lipid peroxidation and oxidative stress. Both groups of LBP treatment significantly reduced or even abolished such elevation of oxidative stress markers (Figures 4H and 4I).
Treatments with LBP attenuated hepatic inflammation and apoptosis through NF-κB
The main pathological discrepancy between steatosis and NASH is the occurrence of hepatic inflammation. To address the anti-inflammatory effect of LBP, we selected three pro-inflammatory markers (TNF-α, IL-1β and COX-2) and one chemokine marker (MCP-1) in all groups of rats for ELISA assays. As expected, the expressions of these proteins were significantly elevated by NASH induction, which were either abolished or significantly attenuated by the co-treatment or delayed-treatment with LBP (Figures 5A–5D). Consistently, hepatic level of cleaved caspase-3, the direct marker for apoptosis, was increased in the NASH rats, indicating the occurrence of hepatic apoptosis. Administrations of LBP effectively reduced its level (Figure 5G). To further explore the mechanism, we tested the expressional change of the intrinsic apoptotic pathway. We found that progression of NASH increased the protein level of cytochrome c and the mRNA level of pro-apoptotic molecule Bax, while decreased in the mRNA level of anti-apoptotic molecule Bcl-2. Both groups of LBP treatment showed significantly reversal of such effects (Figures 5G–5I). Since transcription factor NF-κB is the master regulator of both inflammatory and apoptotic responses, we then measured the activity of its subunit p65 in the nuclear protein. The activity of p65 in NASH group was significantly higher than that of the control group (Figure 5F), indicating an activated status of NF-κB. It was accompanied by a low level of IκBα, the cytosolic inhibitor of NF-κB (Figure 5E). Addition of LBP markedly reduced NF-κB activity and restored the level of IκBα.

LBP therapeutically ameliorated hepatic inflammatory responses and apoptosis through modulating autophagy and MAPK pathway.
(a–d) Protein expressional changes of inflammatory and chemoattractive responses markers tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), cyclooxygenase-2 (COX-2) and monocyte chemotactic protein-1 (MCP-1) from each group of rats after 12-week experiment. (e) Representative Western blot results of inhibitor of NF-κB alpha (IκBα) formation in the liver of each group of rats from 3 repeated Western blot experiments. Full-length blots are presented in Supplementary Fig. 4. (f) Changes of nuclear factor-kappa B (NF-κB) subunit p65 in each group of rats, as measured by EIA kit. (g) Representative Western blot results of cleaved caspase-3 and cytochrome c (cyto c) in rats with different treatments from 3 repeated Western blot experiments. Full-length blots are presented in Supplementary Fig. 4. (h,i) Changes of mRNA expression level of intrinsic apoptotic markers Bcl-2 and Bax in each group of rats. (j) Representative Western blot results of key autophagic markers including phosphorylated mTOR, total mTOR, beclin 1, Atg5, LC3II and p62 in each group of rats from 3 repeated Western blot experiments. Full-length blots are presented in Supplementary Fig. 4. (k) Representative Western blot results of key MAPK markers including phosphorylated and total forms of p38 MAPK, JNK and ERK1/2 in each group of rats from 3 repeated Western blot experiments. Full-length blots are presented in Supplementary Fig. 4. Beta-actin was used as the internal control. Data from each group were expressed as means ± SEMs (n = 5 or 6). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. Different letters (e.g. a and b) mean a statistical significant change (p < 0.05) between each other. C: control, N: NASH, L: vehicle-LBP, NL: NASH + LBP treatment throughout the experiment, TL: vehicle-LBP treatment from the 9th week to the 12th week with NASH induction, NTL: NASH + LBP treatment from the 9th week to the 12th week with NASH induction.
LBP treatments enhanced autophagy and regulated the MAPK pathways
Since autophagy is beneficial for delaying the progression of NASH7, we measured several key autophagic markers in all rats. It is shown that NASH induction in the rat liver up-regulated the expression of autophagic markers including Atg5 and LC3 II, with down-regulation of autophagic negative regulators including phosphorylated mTOR and p62. The influence of NASH on beclin-1 expression was not obvious (Figure 5J). In both groups of LBP treatment (NASH-LBP 12 weeks and NASH-LBP 9-12 weeks), the cellular autophagy was further potentiated, when compared to their expressions in the NASH group (Figure 5J). Vehicle-LBP treatments did not obviously affect the autophagic markers in the healthy rats.
To further investigate the therapeutic property of LBP, we measured three key members (p38 MAPK, JNK and ERK1/2) of the MAPK family. In the NASH group, the phosphorylation levels of both p38 MAPK and JNK were increased, but then decreased to the control level by the administration of LBP (Figure 5K). The phosphorylation of ERK1/2, in contrast, was down-regulated by NASH induction. Both groups of LBP treatment showed further reduction in its phosphorylation level without affecting its total level (Figure 5K).
L-arabinose and β-carotene partially accounted for the hepatic therapeutic effects of LBP
To demonstrate the active monomer(s) that account(s) for the hepatic therapeutic properties of LBP against NASH development, we applied an in vitro cellular steatosis model using sodium palmitate (SP) as the inducer in rat hepatocyte cell line BRL-3A26. After 24-hr incubation, it was found that SP caused evident fat accumulation, with reduced cell viability in a dose-dependent manner (Supplementary Figure 3). Considering the balance between obvious fat accumulation and acceptable cell viability, we chose 0.35 mM SP as the optimum dose in the following in vitro studies. As shown in Figure 6B, application of SP significantly reduced the cell viability from ~100% to ~72%. Co-treatment with LBP restored the cell viability comparable to the control level (~95%). Although not as potent as LBP, treatment with l-arabinose or β-carotene also significantly recovered the cell status to ~84% or ~85%, respectively. In addition, incubation with SP also caused fat accumulation in the cultured BRL-3A cells, which was significantly attenuated by co-treatment with LBP, either with l-arabinose or β-carotene (Figures 6A and 6C). These treatments alone did not influence the normal cells (Figures 6A–6C).

LBP, l-arabinose and β-carotene prevented cellular steatosis and insulin/glucose metabolism dysfunction in a palmitate acid-induced cell model.
(a) Representative image of sodium palmitate-induced fat accumulation in rat hepatocyte cell line BRL-3A after 24-hr incubation. Fat droplets were stained by Oil Red O. Magnification: ×100. Bar: 10 μm. (b) Cellular viability measured by MTT conversion in BRL-3A cells with different treatment for 24 hrs. (c) Quantitative Oil Red O (normalized by MTT results) in BRL-3A cells with different treatment for 24 hrs. In addition, (d) Glucose production and (e) change of the resistin, IRS-1/GSK3α pathways were measured in BRL-3A cells with different treatment for 24 hrs. Data from each group were expressed as means ± SEMs (n = 5). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. Different letters (e.g. a and b) mean a statistical significant change (p < 0.05) between each other.
To further characterize the beneficial roles of l-arabinose and β-carotene, we assessed the insulin and glucose metabolism pathways of the cell. Exposure of BRL-3A cells to SP enhanced gluconeogenesis, which was reduced by LBP or β-carotene treatment (Figure 6D). Western blot results of resistin and phosphorylated IRS-1 further confirmed that SP incubation significantly induced the up-regulation of resistin and the impairment of IRS-1 signaling. Addition of LBP, l-arabinose or β-carotene partially counter-acted these effects. Total protein level of IRS-1 was not influence by these treatments (Figure 6E). In addition, SP incubation caused the increased level of GSK3α phosphorylation, which was partially reduced by the co-treatment of LBP or β-carotene, without affecting its total protein level (Figure 6E). These data indicated that SP incubation induced impaired insulin and glucose metabolism in BRL-3A cells. LBP, l-arabinose and β-carotene, partly ameliorated such impairments.
Similar to the results from the animal model, incubation with SP significantly reduced the expression of CAT and increased the level of TNF-α and cellular apoptosis. Changes of cellular caspase-3/7 activity further proved these findings (Figures 7A–7D). LBP, l-arabinose or β-carotene treatment either abolished or significantly counteracted the effects of SP, although in most cases, LBP showed better alleviative effects than the other two monomers (Figures 7A–7D).

LBP, l-arabinose and β-carotene attenuated cellular oxidative stress, inflammation and apoptosis induced by sodium palmitate.
(a) Catalase (CAT) mRNA, (b) Tumor necrosis factor-alpha (TNF-α), (c) Apoptotic ratio (apoptotic cell number against total cell number) and (d) Cellular caspase-3/7 activity were measured in BRL-3A cells with different treatment for 24 hrs. Data from each group were expressed as means ± SEMs (n = 5). Statistical comparisons between groups were done using the Kruskal–Wallis test followed by Dunn's post hoc test to detect differences in all groups. Different letters (e.g. a and b) mean a statistical significant change (p < 0.05) between each other.
Discussion
Since the initiation and progression of NAFLD/NASH is a slow and progressive process, using an appropriate therapy that can target most of its key pathological events (e.g. insulin resistance, lipid deposition, fibrosis and inflammation) and combined with specific exercise plan, should theoretically reverse or retard the process of fatty liver to a nearly healthy condition4,27. Here we showed that LBP could therapeutically attenuate a spectrum of NASH phenotypes, from up-stream insulin resistance to down-stream hepatic apoptosis in both in vivo and in vitro systems.
Insulin resistance and glucose metabolism impairment are found in most NAFLD/NASH patients. It is characterized by a reduce insulin sensitivity in the whole body, liver and adipose tissues. In the liver, it mainly causes excess dietary fat, enhanced delivery of free fatty acids to the liver, diminished fatty acid oxidation and increased de novo lipogenesis28,29. In this study, from GTT (with both normalized glucose and insulin levels) and ITT readouts, both groups of LBP administration (NASH-LBP × 12 weeks and post-NASH/LBP × 4 weeks) potently improved insulin sensitivity and glucose metabolism in the NASH rats (Figure 1 and Supplementary Figure 2). Therapeutic LBP treatment exhibited comparable ameliorative effects as with the LBP co-treated rats. In NASH rats, apart from the above changes, this was accompanied by the impaired hepatic signaling of IRS-1 and activated signaling of GSK3α, key molecules in insulin resistance and glucose metabolism, respectively30,31. Furthermore, the excessive hepatic fat accumulation and serum FFA enhancement were significantly attenuated by LBP treatments. Molecular markers for fatty acid biosynthesis and utilization also revealed the re-balancing of the lipid metabolism by LBP (Figs. 2 and 4). These results were consistent with our previous findings15 and other reports showing the beneficial effects of LBP on steatosis32,33.
Fibrosis is one of the major features of fatty liver diseases. Activation of HSCs and following PI3K/Akt/FoxO1 pathway contributes to the initiation of fibrosis, with well-characterized TGF-β/SMAD pathway34,35. We found that high-fat diet significantly enhanced the collagen formation in the liver (Figure 2), which was accompanied by elevated levels of the TGF-β/SMAD pathway and HSCs activation (Figure 3). When LBP was administered along with or after the establishment of NASH, it evidently ameliorated the formation of hepatic collagen through inhibition of these signaling pathways. Thus, LBP may share similar mechanistic pathways with other hepato-protective herbal derivatives, such as green tea on fibrotic attenuation8.
It is well-known that NF-κB is a major sensor and regulator of oxidative stress, inflammation and apoptosis36,37. The activation of NF-κB relies on the degradation of its cytosolic inhibitor IκBα. In the current study, high-fat diet induced NASH rats showed enhanced activity of p65 and promoted the degradation of IκBα, indicating an activated status of NF-κB, accompanied with the occurrence of oxidative stress, inflammation and apoptosis in the liver (Figures 4 and 5). Similar with our previous studies using garlic, green tea extract and LBP itself6,8,15, supplementation of LBP, either protectively or therapeutically, significantly alleviated hepatic oxidative stress, inflammatory response and apoptosis during NASH development partially through NF-κB inhibition. Since dysregulated lipid metabolism may induce lipid peroxidation, directly leading to oxidative stress, inflammation and apoptosis, the beneficial function of LBP on lipid metabolism may also partly explain the attenuated hepatic injury by LBP treatment38.
In recent years, the beneficial property of autophagy in NAFLD/NASH development drew much attention because it counteracted the effect of apoptosis and also hepato-protective drug treatment usually enhances the activity of autophagy, as we demonstrated in the garlic study7. A recent paper using ULK1 knockout mice also pointed out that impairment of the autophagic signaling significantly influenced the therapeutic effects of resveratrol on NAFLD mice39. In accordance to these studies, our two approaches of LBP treatment also enhanced the autophagic pathway activity (including beclin 1, Atg 5 and LC3II) through the degradation of mTOR, when compared with the NASH rat group. Furthermore, the expression level of autophagic substrate p62 was down-regulated by NASH development and further decreased by treatments with LBP, indicating the enhanced autophagy in the liver from another aspect40.
Similar to most Traditional Chinese Medicine recipes, LBP is a mixture of several monomers including glucose, arabinose, galactose, mannose, xylose, rhamnose and/or fucose41,42. It also contains other antioxidants such as zeaxanthin dipalmitate, β-cryptoxanthin palmitate, zeaxanthin monopalmitate and β-carotene43,44. Previous studies have extensively examined the beneficial properties of LBP on tumor, neuronal system and the liver. However, none of them characterized the effective components of LBP. In the current study, we firstly found that l-arabinose and β-carotene partially represented the active functional components of LBP in lipid, insulin and glucose metabolism, oxidative stress, inflammation and apoptosis in a palmitate acid-induced cellular steatosis model (Figures 6 and 7). Although LBP showed stronger protection than l-arabinose or β-carotene, our current results demonstrated the active beneficial functions of its components for the possible treatment of liver diseases such as NAFLD and NASH.
Together, our findings showed a potent therapeutic and hepato-protective effect of LBP in ameliorating a spectrum of NASH induced hepatic abnormalities, including (1) increased body and liver weights; (2) hepatic insulin resistance and glucose metabolism dysfunction; (3) elevated level of serum aminotransferases; (4) fat accumulation in the liver and increased serum FFA level; (5) hepatic fibrosis; (6) hepatic oxidative stress; (7) hepatic inflammatory response; and (8) hepatic apoptosis. These improvements were partially through the modulation of transcription factor NF-κB, MAPK pathways and the autophagic processes. Importantly, LBP not only alleviated hepatic injury when administered along with the induction of NASH, but also therapeutically recovered liver functions on an established NASH condition. In vitro studies exhibited the beneficial properties of LBP partly ascribed to the functions of l-arabinose and β-carotene. Thus, daily consumption of LBP should be considered as a promising way to prevent and retard the initiation and progression of NASH.
References
Browning, J. D. et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 40, 1387–1395 (2004).
Michelotti, G. A., Machado, M. V. & Diehl, A. M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 10, 656–665 (2013).
Yilmaz, Y. Review article: is non-alcoholic fatty liver disease a spectrum, or are steatosis and non-alcoholic steatohepatitis distinct conditions? Aliment. Pharmacol. Ther. 36, 815–823 (2012).
Xiao, J., Guo, R., Fung, M. L., Liong, E. C. & Tipoe, G. L. Therapeutic approaches to non-alcoholic fatty liver disease: past achievements and future challenges. Hepatobiliary Pancreat. Dis. Int. 12, 125–135 (2013).
Vitaglione, P. et al. Coffee reduces liver damage in a rat model of steatohepatitis: the underlying mechanisms and the role of polyphenols and melanoidins. Hepatology. 52, 1652–1661 (2010).
Xiao, J. et al. Garlic-derived S-allylmercaptocysteine is a hepato-protective agent in non-alcoholic fatty liver disease in vivo animal model. Eur. J. Nutr. 52, 179–191 (2013).
Xiao, J. et al. Garlic-Derived S-Allylmercaptocysteine Ameliorates Nonalcoholic Fatty Liver Disease in a Rat Model through Inhibition of Apoptosis and Enhancing Autophagy. Evid. Based Complement. Alternat. Med. 2013, 642920 (2013).
Xiao, J. et al. Epigallocatechin gallate attenuates fibrosis, oxidative stress and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1 and NF-kappa B pathways. Eur. J. Nutr. 10.1007/s00394-013-0516-8 (2013).
Cacciapuoti, F., Scognamiglio, A., Palumbo, R., Forte, R. & Cacciapuoti, F. Silymarin in non alcoholic fatty liver disease. World J. Hepatol. 5, 109–113 (2013).
Xin, P. et al. Alleviative effects of resveratrol on nonalcoholic fatty liver disease are associated with up regulation of hepatic low density lipoprotein receptor and scavenger receptor class B type I gene expressions in rats. Food Chem. Toxicol. 52, 12–18 (2013).
Chang, R. C. & So, K. F. Use of anti-aging herbal medicine, Lycium barbarum, against aging-associated diseases. What do we know so far? Cell. Mol. Neurobiol. 28, 643–652 (2008).
Zhang, M. et al. Effect of lycium barbarum polysaccharide on human hepatoma QGY7703 cells: inhibition of proliferation and induction of apoptosis. Life Sci. 76, 2115–2124 (2005).
Li, S. Y. et al. Lycium barbarum polysaccharides reduce neuronal damage, blood-retinal barrier disruption and oxidative stress in retinal ischemia/reperfusion injury. PloS one 6 (2011).
Xiao, J. et al. Lycium barbarum polysaccharides protect mice liver from carbon tetrachloride-induced oxidative stress and necroinflammation. J. Ethnopharmacol. 139, 462–470 (2012).
Xiao, J. et al. Lycium barbarum polysaccharides protect rat liver from non-alcoholic steatohepatitis-induced injury. Nutr. Diabetes. 3, e81 (2013).
Tipoe, G. L. et al. Voluntary oral feeding of rats not requiring a very high fat diet is a clinically relevant animal model of non-alcoholic fatty liver disease (NAFLD). Histol. Histopathol. 24, 1161–1169 (2009).
Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 41, 1313–1321 (2005).
Cheng, K. K. et al. APPL1 potentiates insulin-mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab. 9, 417–427 (2009).
McGuinness, O. P., Ayala, J. E., Laughlin, M. R. & Wasserman, D. H. NIH experiment in centralized mouse phenotyping: the Vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. Am. J. Physiol. Endocrinol. Metab. 297, E849–E855 (2009).
Krog-Mikkelsen, I. et al. The effects of L-arabinose on intestinal sucrase activity: dose-response studies in vitro and in humans. Am. J. Clin. Nutr. 94, 472–478 (2011).
Martin, K. R., Failla, M. L. & Smith, J. C., Jr Beta-carotene and lutein protect HepG2 human liver cells against oxidant-induced damage. J. Nutr. 126, 2098–2106 (1996).
Xiao, J. et al. Tumor necrosis factor-alpha gene from mandarin fish, Siniperca chuatsi: molecular cloning, cytotoxicity analysis and expression profile. Mol. Immunol. 44, 3615–3622 (2007).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009).
Cassiman, D. et al. Synaptophysin: A novel marker for human and rat hepatic stellate cells. Am. J. Pathol. 155, 1831–1839 (1999).
Albano, E., Mottaran, E., Occhino, G., Reale, E. & Vidali, M. Review article: role of oxidative stress in the progression of non-alcoholic steatosis. Aliment. Pharmacol. Ther. 22 Suppl 2, 71–73 (2005).
Zhang, Y. et al. Silibinin ameliorates steatosis and insulin resistance during non-alcoholic fatty liver disease development partly through targeting IRS-1/PI3K/Akt pathway. Int. Immunopharmacol. 17, 714–720 (2013).
Mahady, S. E. & George, J. Management of nonalcoholic steatohepatitis: an evidence-based approach. Clin. Liver Dis. 16, 631–645 (2012).
Polyzos, S. A., Kountouras, J. & Zavos, C. Nonalcoholic fatty liver disease: the pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 9, 299–314 (2009).
Utzschneider, K. M. & Kahn, S. E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 91, 4753–4761 (2006).
Draznin, B. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: the two sides of a coin. Diabetes 55, 2392–2397 (2006).
Ciaraldi, T. P., Nikoulina, S. E., Bandukwala, R. A., Carter, L. & Henry, R. R. Role of glycogen synthase kinase-3 alpha in insulin action in cultured human skeletal muscle cells. Endocrinology 148, 4393–4399 (2007).
Cheng, D. & Kong, H. The effect of Lycium barbarum polysaccharide on alcohol-induced oxidative stress in rats. Molecules. 16, 2542–2550 (2011).
Ming, M., Guanhua, L., Zhanhai, Y., Guang, C. & Xuan, Z. Effect of the Lycium barbarum polysaccharides administration on blood lipid metabolism and oxidative stress of mice fed high-fat diet in vivo. Food Chem. 113, 872–877 (2009).
Feldstein, A. E. et al. Hepatic stellate cells and fibrosis progression in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 3, 384–389 (2005).
Adachi, M. et al. The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells. Gastroenterology. 132, 1434–1446 (2007).
van den Berg, R., Haenen, G. R., van den Berg, H. & Bast, A. Transcription factor NF-kappaB as a potential biomarker for oxidative stress. Br. J. Nutr. 86 Suppl 1, S121–127 (2001).
Wang, H. & Cho, C. H. Effect of NF-kappaB signaling on apoptosis in chronic inflammation-associated carcinogenesis. Curr. Cancer Drug Targets. 10, 593–599 (2010).
Sumida, Y., Niki, E., Naito, Y. & Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 47, 869–880 (2013).
Li, L. et al. Resveratrol modulates autophagy and NF-kappaB activity in a murine model for treating non-alcoholic fatty liver disease. Food Chem. Toxicol. 63, 166–173 (2014).
Moscat, J. & Diaz-Meco, M. T. p62 at the crossroads of autophagy, apoptosis and cancer. Cell. 137, 1001–1004 (2009).
Li, X. L., Zhou, A. G. & Li, X. M. Inhibition of Lycium barbarum polysaccharides and Ganoderma lucidum polysaccharides against oxidative injury induced by γ-irradiation in rat liver mitochondria. Carb. Pol. 69, 172–178 (2007).
Wang, C. C., Chang, S. C. & Chen, B. H. Chromatographic determination of polysaccharides in Lycium barbarum Linnaeus. Food Chem. 116, 595–603 (2009).
Inbaraj, B. S. et al. Determination of carotenoids and their esters in fruits of Lycium barbarum Linnaeus by HPLC-DAD-APCI-MS. J. Pharm. Biomed. Anal. 47, 812–818 (2008).
Peng, Y. et al. Quantification of zeaxanthin dipalmitate and total carotenoids in Lycium fruits (Fructus Lycii). Plant Foods Hum. Nutr. 60, 161–164 (2005).
Acknowledgements
This work was supported by Small Project Funding, University Research Committee, HKU, General Research Fund, University Grant Council, Hong Kong SAR, Azalea (1972) Endowment Fund to K.F.S. and R.C.C.C., Health Medical Research Fund, HKSAR and Fundamental Research Funds for the Central Universities (No. 21609101) to K.F.S.
Author information
Authors and Affiliations
Contributions
J.X., K.F.S. and G.L.T. designed the study. J.X., F.X., J.H. and E.C.L. performed the experiments. M.L.F., Y.P.C., R.C.C.C. and A.X. offered LBP, essential experimental materials and expertise on molecular biology techniques. J.X., M.L.F., Y.P.C., R.C.C.C., A.X., K.F.S. and G.L.T. analyzed the data. J.X., F.X., M.L.F., E.C.L., K.F.S. and G.L.T. wrote and edited the manuscript. All authors reviewed and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary data all-in-one
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Xiao, J., Xing, F., Huo, J. et al. Lycium barbarum polysaccharides therapeutically improve hepatic functions in non-alcoholic steatohepatitis rats and cellular steatosis model. Sci Rep 4, 5587 (2014). https://doi.org/10.1038/srep05587
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05587
This article is cited by
-
Fermented mulberry (Morus alba) leaves suppress high fat diet-induced hepatic steatosis through amelioration of the inflammatory response and autophagy pathway
BMC Complementary Medicine and Therapies (2020)
-
Co-stimulation of LPAR1 and S1PR1/3 increases the transplantation efficacy of human mesenchymal stem cells in drug-induced and alcoholic liver diseases
Stem Cell Research & Therapy (2018)
-
Lycium barbarum polysaccharide protects HSF cells against ultraviolet-induced damage through the activation of Nrf2
Cellular & Molecular Biology Letters (2018)
-
Targeting autophagy in obesity: from pathophysiology to management
Nature Reviews Endocrinology (2018)
-
Sulfated Cyclocarya paliurus polysaccharides markedly attenuates inflammation and oxidative damage in lipopolysaccharide-treated macrophage cells and mice
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
