
Abstract
Study design: Case report.
Objective: We describe a patient who developed a myelopathy associated with a noncompressive herniated cervical intervertebral disc at the same level. We provide clinical and radiological evidence that reveals that even though the disc herniation did not compress the spinal cord, it diminished venous blood flow out of the spinal cord, possibly resulting in a venous hypertensive myelopathy (VHM).
Setting: Baltimore, MD, USA.
Clinical presentation: A 29-year-old woman developed a cervical radiculopathy, followed by a slowly progressive cervical myelopathy associated with a herniated C5–C6 disc. Magnetic resonance imaging showed a noncompressive disc herniation, a swollen spinal cord with increased T2 signal most prominent at the site of the herniated disc, extending several levels above and below the disc. The patient was diagnosed with acute transverse myelitis (ATM) and was started on IV steroids. However, unlike most cases of transverse myelitis, spinal fluid analysis was noninflammatory. In contrast, several features suggested that the patient instead had VHM. We suggest that the disc herniation resulted in impaired drainage of blood from the spinal cord through compression of the venous plexus near the intervertebral foramen.
Intervention: Although the patient did not recover function following high-dose steroid administration, she recovered completely following C5–C6 discectomy and fusion.
Conclusion: To our knowledge, this is the first report of likely VHM in the absence of a spinal arteriovenous malformation. We suggest that some patients diagnosed with ATM in the setting of extrinsic spinal column abnormalities may actually have a noninflammatory myelopathy associated with impaired spinal venous drainage.
Similar content being viewed by others


Introduction
Acute transverse myelitis (ATM) is a group of disorders characterized by focal inflammation of the spinal cord and resultant neural dysfunction and injury. ATM may be an isolated entity or may occur in the context of multifocal or even multisystemic disease. Recently, we have proposed a diagnostic and classification scheme, which has defined ATM either as idiopathic or associated with a known inflammatory disease (ie multiple sclerosis, systemic lupus erythematosus, Sjogren's syndrome, or neurosarcoidosis).1 ATM is characterized by acutely developing symptoms and signs of neurological dysfunction in motor, sensory and autonomic nerves, and nerve tracts of the spinal cord. There is often a clearly defined rostral border of sensory dysfunction and a spinal magnetic resonance imaging (MRI) and lumbar puncture often show evidence of acute inflammation.2,3,4,5,6,7,8 When the maximal level of deficit is reached, approximately 50% of patients have lost all movements of their legs, virtually all patients have some degree of bladder dysfunction, and 80–94% of patients have numbness, paresthesias or band-like dysesthesias.5,6,7,8,9,10 Autonomic symptoms consist variably of increased urinary urgency, bowel or bladder incontinence, difficulty or inability to void, incomplete evacuation or bowel constipation.11
An alternative form of noncompressive transverse myelopathy may be caused by vascular abnormalities in or near the spinal cord. The distinction between inflammatory and vascular pathophysiology may be difficult, leading to improper diagnosis and therapy. Venous drainage of the spinal cord occurs via both intrinsic and extrinsic spinal cord veins. The intrinsic spinal cord veins consist of an anterior median group and a radial group.12 Blood in the anterior third of the spinal cord drains to the segmental central vein, then to the longitudinally oriented anterior median spinal vein, which lies on the pial surface of the spinal cord in the midline. The radial veins form in the peripheral gray matter or white matter and drain radially to the coronal plexus of veins on the pial surface. The anterior median spinal vein and the coronal plexus (termed the extrinsic venous drainage) are drained by the medullary veins that travel with nerve roots. In the intervertebral foramen, a number of veins including the anterior and posterior medullary veins, veins from the vertebral plexus and the radicular veins coalesce to form a plexus surrounding the spinal nerve.
Dural arteriovenous malformations (AVMs) have been reported to cause neurologic dysfunction on the basis of venous hypertension.13,14,15,16 Surgical studies have shown that interruption of the shunt halts progression and can lead to functional improvement in a substantial proportion of patients, implying that the clinical dysfunction was due to venous congestion.17 Pathological studies of spinal tissue associated with a spinal dural arteriovenous fistula (AVF) have shown marked white matter hypocellularity and small vessels with thickened, hyalinized walls. Extensive vascular sclerosis and gliosis of the white matter was demonstrated with disorganization of the white matter, patchy myelin loss, and concomitant axonal loss. These findings were similar to those found in an ischemic myelopathy secondary to venous congestion, first described by Foix and Alajouanine.18
Case report
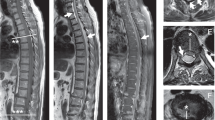
The patient is a 29-year-old white female, who first noted tingling in her right hand after doing some yard work. She developed progressive weakness in the right arm, and physical examination 2 months later showed mild weakness in the right bicep, diminished bicep reflex, but an otherwise normal neurologic exam. Electrophysiologic examination of the involved extremity was normal. MRI of the cervical spine showed herniation of the C5–C6 disc, which flattened the thecal sac at that level and encroached the right neuroforamen at that level (Figure 1a, sagittal-top; axial-bottom). There was no spinal cord compression and there was preservation of the anterior rim of cerebrospinal fluid (CSF) on all axial images. Flexion-extension X-rays of the neck did not show any hypermobility or frank subluxation. Repeat sagittal MRI images of the neck with the patient in the flexed and extended position did not reveal a positional compression of the spinal cord. Surgery was recommended, however, because of the right-sided radiculopathy. While the patient was considering surgery, she developed clumsiness and weakness in her right lower extremity, a band-like sensation around her chest above her breasts, loss of sensation in both hands, and ‘stiffness’ in the lower extremities. These symptoms slowly progressed over a 4–6 week period, causing her to seek medical attention. Physical examination at that point showed diminished fine motor control in both arms, weakness in both biceps and wrist extensors, and increased tone with brisk reflexes in both lower extremities. Plantar responses were flexor and no truncal sensory level was detected. MRI revealed interval development of diffuse intramedullary increased T2 signal in the spinal cord from C2 to C7 (Figure 1b, arrowhead; sagittal-top; axial-bottom). A peripheral rim of T2 hypointensity was detected (Figure 1b, arrows; sagittal-top; axial-bottom). The spinal cord appeared diffusely swollen with heterogeneous contrast enhancement in the posterior aspect of the spinal cord and a peripheral rim of T2 hypointensity (Figure 1c, arrowhead; sagittal-top, axial-bottom).

(a) T2 sagittal (top) and axial (bottom) MRI 6 weeks after the development of symptoms and signs of a right C5 radiculopathy. Note acute disc herniation at C5–C6, encroaching upon the neuroforamen at that level and no spinal cord compression. (b) T2 sagittal (top) and axial (bottom) MRI after the development of symptoms and signs of a cervical myelopathy. Note interval development of intramedullary increased T2 signal (arrowheads) and peripheral rim of T2 hypointensity (arrows). (c) T1 sagittal (top) and axial (bottom) MRI with gadolinium after the development of symptoms and signs of a cervical myelopathy. Note gadolinium enhancement in posterior aspect of the spinal cord. (d) T2 sagittal (top) and axial (bottom) MRI 6 weeks after C5–C6 anterior cervical discectomy with osteophytectomy and allograft arthodesis with spinal instrumentation. Note complete resolution of signal abnormality
The patient was diagnosed with transverse myelitis and started on intravenous methylprednisolone. Laboratory analysis revealed normal CBC, electrolytes, B12, and folate level. Heavy metal screen and hepatitis serology were negative. SSA, SSB, cryoglobulins, ESR, and ANA were unremarkable. Spinal fluid analysis, performed prior to the institution of methylprednisolone and while symptoms were continuing to worsen, was totally acellular with normal protein levels, absent oligoclonal bands, and a normal IgG index. The patient did not respond to steroid infusions, and examination 1 month later was unchanged from previous examinations.
The patient underwent C5–C6 anterior cervical discectomy with osteophytectomy and allograft arthodesis with spinal instrumentation. She did well following surgery with resolution of the band-like sensation and stiffness in her lower extremities by the time of hospital discharge 3 days later. Follow-up examination 3 months following the operation revealed normal tone and muscle strength throughout, although fine motor control remained diminished in her lower extremities. Deep tendon reflexes normalized with resolution of pathologic reflexes. Follow-up MRI of the cervical spinal cord revealed complete resolution of the described abnormalities (Figure 1d, sagittal-top and axial-bottom).
Discussion
We describe a patient who developed a subacute cervical myelopathy temporally following herniation of a cervical disc. The herniated disc caused neuroforaminal encroachment, but did not compress the spinal cord. Her physicians determined that the herniated disc was unrelated to the developing myelopathy, and the patient was diagnosed with ATM and was treated with intravenous steroids.
There are several reasons why we believe that this patient did not have ATM. First, unlike most patients with ATM, this patient had no evidence of inflammation in the CSF. Second, no evidence of an underlying inflammatory disorder was found. Third, unlike virtually all patients with ATM,4,6,8,9,10,11 the patient in the current report never developed any autonomic dysfunction. Fourth, the patient did not respond to high-dose intravenous steroids. Finally, the clinical progression of her myelopathy occurred over a 4–6 week period, which is slower than that seen in ATM and indeed is an exclusionary criterion according to recently published guidelines for the diagnosis of ATM.1
Therefore, we suggest that this myelopathy may instead be due to venous hypertensive myelopathy (VHM). Although the evidence remains somewhat circumstantial, there were several clinical and radiologic features that support this notion. First, the development of intramedullary signal abnormality in the spinal cord with maximal involvement centered at the level of the disc herniation suggested a link between the two. Second, several imaging characteristics seen in the current patient have been described in patients with VHM. Elevated T2 abnormality, often occupying the central portion of the spinal cord, gadolinium enhancement in a linear array on sagittal images and a peripheral rim of T2 hypointensity have all been reported in patients with VHM secondary to dural AVF.19,20,21,22,23 The peripheral T1 hypointensity may appear as a dark rim around the periphery of the spinal cord and can be seen on sagittal and axial images. This rim of diminished T2 signal may represent slow flow of blood containing deoxyhemoglobin within the spinal cord or accumulation of paramagnetic substances within the spinal cord proper secondary to leakage from damaged vessels. Gadolinium enhancement may represent disruption of the blood–brain barrier secondary to venous infarct or ischemia and may occur in a linear manner (rather than irregular or ring enhancement with ATM). Third, the patient had a rapid and complete response to surgical decompression. Fourth, slowly progressive myelopathic symptoms over 4–6 weeks are more consistent with a stuttering myelopathy associated with VHM rather than inflammation. Finally, resolution of the imaging abnormality is more likely to occur following VHM, rather than persistence of T2 abnormality due to a gliotic/demyelinated area in ATM.23 We suggest that diminished venous return, likely of anterior and posterior medullary veins draining through the right intervertebral foramina at C5–C6, resulted in decreased venous return from the spinal cord and caused the development of VHM. It is likely that the venous plexus was compressed just prior to the intervertebral foramen. The veins draining the spinal cord to this point are valveless and therefore, effectively transmit backflow of venous drainage into the spinal cord. Conversely, veins at or distal to the junction of the medullary veins and the vertebral plexus have valves and, therefore, compression at or distal to the intervertebral foramina would result in limited backflow of venous blood into the spinal cord.24
Neurologic symptoms in patients with VHM are likely a result of increased venous pressure, with subsequent increase in intramedullary arterial vasodilation in order to maintain perfusion pressure. These changes then result in increased tissue pressure, possibly associated with progressive exhaustion of the autoregulatory capacity.25 Ultimately, intramedullary edema develops, leading to a decrease in perfusion pressure and ischemic injury. Although the mechanism of VHM in the setting of arteriovenous malformations is different (ie a direct communication between arteries and veins with increased venous pressure as a result), the pathophysiologic sequelae may be similar to that with a venous obstructive lesion.
There is no way to know how commonly VHM occurs (in the setting of intervertebral disc herniation or vertebral compression fracture, for example), and we suggest that every patient with evolving myelopathy receive a workup to define structural, metabolic or inflammatory causes. However, in many of the reported patients with VHM, a slowly progressive myelopathy developed into a severe necrotizing myelopathy, likely the result of progressive elevation in venous pressure and subsequent ischemic injury with or without venous thrombosis. Therefore, VHM should be considered in any patient with an evolving noncompressive myelopathy and the clinical and radiologic characteristics described above.
References
Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology 2002; 59: 499–505.
Scott TF, Bhagavatula K, Snyder PJ, Chieffe C . Transverse myelitis. Comparison with spinal cord presentations of multiple sclerosis. Neurology 1998; 50: 429–433.
Al Deeb SM, Yaqub BA, Bruyn GW, Biary NM . Acute transverse myelitis. A localized form of postinfectious encephalomyelitis. Brain 1997; 120 (Part 7): 1115–1122.
Ropper AH, Poskanzer DC . The prognosis of acute and subacute transverse myelopathy based on early signs and symptoms. Ann Neurol 1978; 4: 51–59.
Lipton HL, Teasdall RD . Acute transverse myelopathy in adults. A follow-up study. Arch Neurol 1973; 28: 252–257.
Berman M et al. Acute transverse myelitis: incidence and etiologic considerations. Neurology 1981; 31: 966–971.
Christensen PB, Wermuth L, Hinge HH, Bomers K . Clinical course and long-term prognosis of acute transverse myelopathy. Acta Neurol Scand 1990; 81: 431–435.
Jeffery DR, Mandler RN, Davis LE . Transverse myelitis. Retrospective analysis of 33 cases, with differentiation of cases associated with multiple sclerosis and parainfectious events. Arch Neurol 1993; 50: 532–535.
Misra UK, Kalita J, Kumar S . A clinical MRI and neurophysiological study of acute transverse myelitis. J Neurol Sci 1996; 138: 150–156.
Altrocchi PH . Acute transverse myelopathy. Arch Neurol 1963; 9: 21–29.
Sakakibara R, Hattori T, Yasuda K, Yamanishi T . Micturition disturbance in acute transverse myelitis. Spinal Cord 1996; 34: 481–485.
Gillilan LA . Veins of the spinal cord. Anatomic details; suggested clinical applications. Neurology 1970; 20: 860–868.
Symon L, Kuyama H, Kendall B . Dural arteriovenous malformations of the spine. Clinical features and surgical results in 55 cases. J Neurosurg 1984; 60: 238–247.
Shephard RH . Observations on intradural spinal angioma: treatment by excision. Neurochirurgia (Stuttg) 1963; 6: 58–74.
Pia HW . Diagnosis and treatment of spinal angiomas. Acta Neurochir 1973; 28: 1–12.
Aminoff MJ, Barnard RO, Logue V . The pathophysiology of spinal vascular malformations. J Neurol Sci 1974; 23: 255–263.
Oldfield EH et al. Successful treatment of a group of spinal cord arteriovenous malformations by interruption of dural fistula. J Neurosurg 1983; 59: 1019–1030.
Foix C, Alajouanine T . La myelite nécrotique subaiguë. Myelite centrale angio-hipertrophique a évolution progressive. Paraplégie amyotrophique lentement ascendante, d'abord spasmodique puis 15 flasque, s'accompagnantde dissociation albumino-cytologique. Rev Neurol Paris 1926; 33: 1–42.
Bowen BC et al. Spinal dural arteriovenous fistulas: evaluation with MR angiography. AJNR Am J Neuroradiol 1995; 16: 2029–2043.
De Marco JK, Dillon WP, Halback VV, Tsuruda JS . Dural arteriovenous fistulas: evaluation with MR imaging. Radiology 1990; 175: 193–199.
Gilbertson JR, Miller GM, Goldman MS, Marsh WR . Spinal dural arteriovenous fistulas: MR and myelographic findings. AJNR Am J Neuroradiol 1995; 16: 2049–2057.
Isu T, Iwasaki Y, Akino M, Koyanagi I, Abe H . Magnetic resonance imaging in cases of spinal dural arteriovenous malformation. Neurosurgery 1989; 24: 919–923.
Hurst RW, Grossman RI . Peripheral spinal cord hypointensity on T2-weighted MR images: a reliable imaging sign of venous hypertensive myelopathy. AJNR Am J Neuroradiol 2000; 21: 781–786.
Clemens HJ . Die Venensysteme der Menschlichen Wirbelsäule. Walter de Gruyter and Co: Berlin, 1961.
Smith AJ, McCreery DB, Bloedel JR, Chou SN . Hyperemia, CO2 responsiveness, and autoregulation in the white matter following experimental spinal cord injury. J Neurosurg 1978; 48: 239–251.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Krishnan, C., Malik, J. & Kerr, D. Venous hypertensive myelopathy as a potential mimic of transverse myelitis. Spinal Cord 42, 261–264 (2004). https://doi.org/10.1038/sj.sc.3101517
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.sc.3101517
Keywords
This article is cited by
-
Analysis of spinal angiograms that missed diagnosis of spinal vascular diseases with venous hypertensive myelopathy: the non-technical factors
European Spine Journal (2020)
-
Posttraumatic subacute ascending myelopathy in a 24-year-old male patient
Emergency Radiology (2010)
-
Subacute progressive ascending myelopathy following spinal cord injury: MRI appearances and clinical presentation
Spinal Cord (2008)
