
Abstract
Progress has slowed substantially in improving survival rates for pediatric sarcomas, particularly in refractory and metastatic disease. Significant progress has been made in the field of tumor vaccines for such malignancies, which target established tumor antigens. While tumor vaccines have demonstrated safety and improved survival rates, they are inadequate in mediating the regression of established tumor masses and metastases. Programmed cell death ligand 1 (PDL1) is a cell-surface protein induced in a number of adult malignancies. By acting on the corresponding T-cell receptor PD1, PDL1 is able to suppress cytotoxic T-cell–mediated tumor responses. Recent therapeutics blocking this interaction have shown promise in various adult cancers by restoring a functional T-cell response and by directing this response toward an activated, rather than regulatory, T-cell phenotype. We shall discuss the current state of tumor vaccines targeting pediatric sarcomas, review PD1–PDL1 interactions and current therapies targeting these interactions in adult malignancies, and discuss recent studies in which tumor vaccines, combined with PDL1 blockades, produced superior tumor regression compared with the vaccine alone. These studies provide a compelling case for investigation of PDL1 expression and its inhibition in pediatric sarcomas, while continuing to utilize tumor vaccines in tandem to achieve superior clinical outcomes.
Similar content being viewed by others

Introduction
Sarcomas are rare malignancies of the bone and soft tissue, including osteosarcoma, rhabdomyosarcoma, nonrhabdomyosarcoma soft tissue sarcoma (STS), and Ewing’s sarcoma and collectively account for roughly 10% of childhood cancers (1,2). Although considerable progress on survival rates had been made initially with regard to these malignancies, progress has stagnated in the past 25 years (3). Survival rates remain dismal for those patients who present with advanced metastatic disease, recurrence, or those who are refractory to conventional chemotherapy (4,5,6,7,8). Additionally, short-term toxicities from conventional chemotherapy can often hamper the treatment course itself. Long-term toxicities from chemotherapy can manifest in adult survivors as well, and the risk of secondary malignancies looms after a successful clinical outcome (9). Increased-intensity chemotherapeutic regimens for these refractory cancers have failed to yield improved survival rates (10,11,12). Therefore, there remains an urgent need for new treatment modalities that offer both fewer side effects and better clinical outcomes.
Tumor Vaccines in Pediatric Sarcomas
One novel therapeutic concept that has been explored is the development of tumor vaccines. Following the discovery of molecular markers on cancer cells which served as targets for T-cell recognition, a wide variety of tumor vaccines were designed with the goal of achieving a more focused and potent antitumor response to these tumor antigens from host T cells (13). Exact methods for achieving such a response have been widely varied, but more recent trials in pediatric sarcomas have relied on host dendritic cells (DCs) for the stimulation of a primary host immune response via the presentation of tumor antigens (14,15). In such tumor vaccines, autologous DCs are harvested and expanded and are then “pulsed” with tumor lysate ex vivo, or chemically fused with autologous tumor cells themselves (13,15,16,17,18,19,20).
There are currently several ongoing tumor vaccine trials for pediatric sarcomas, which are summarized in Table 1 (13,16). NCT01241162, which is currently recruiting patients, uses a tumor vaccine consisting of autologous DCs pulsed with cancer testis antigen to generate an immune response against Ewing sarcoma, osteosarcoma, rhabdomyosarcoma, and synovial sarcoma. Another trial, NCT00405327, employs tumor lysate-pulsed autologous DCs for the treatment of high-risk solid tumors following stem-cell transplantation. Additionally, NCT00923351 utilizes harvested tumor lysates from pediatric rhabdomyosarcoma and Ewing sarcoma patients, which are then pulsed through autologous DCs with or without recombinant IL-7. In adult and pediatric cancer alike, tumor vaccines have been shown to be consistently safe and effective at slowing tumor growth and improving survival, but only rarely cause regression in established tumors; furthermore, they are not approved for single-agent therapy (13,15,16,21,22,23,24,25). This is likely due to multiple factors that contribute to the phenomenon of immune escape by tumors, which are largely facilitated by suppression of the cytotoxic T-cell response against the tumor (13,17). This suppression can be directly mediated by the tumor itself, or by subpopulations of cells, including myeloid-derived suppressor cells and T regulatory cells (Tregs) which, following their induction by proinflammatory cytokines within the tumor microenvironment, suppress robust immune responses via either antigen-specific or nonspecific manners. Novel means of circumventing this phenomenon of immune escape, either through new mechanisms or further refinement of existing tumor vaccines, are of paramount importance for the improvement of therapeutic options in pediatric tumors.
Targeting the PD1/PDL1 Pathway and its Applicability to Pediatric Cancers
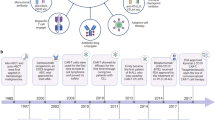
The PD1/PDL1 pathway has recently emerged as a central component of immune regulation that is also utilized by cancer cells to evade host immune response ( Figure 1 ). Under normal conditions, PD1 (also known as BH7) is expressed on activated CD8+ T cells, and its interaction with PDL1 on host tissues leads to the inhibition of T-cell receptor (TCR) signaling via SHP1/2, limits the interactions between T cells and DCs, and ultimately leads to T-cell inactivation and apoptosis (26,27,28,29,30). Additionally, PD1/PDL1 signaling has been shown to convert Th1 cells into FOXP3+ Tregs, thereby preventing clonal T-cell expansion (31). PDL1 can be constitutively expressed or induced by localized inflammatory stimuli, such as interferons (27). This pathway is critical for preventing autoimmunity by maintaining self-tolerance to protect host tissues from any protracted immune response (27).

Initial activation of CD8+ T cells occurs when dendritic cells (DCs) present tumor antigen in context of major histocompatibility class I (MHC I) to the T-cell receptor (TCR). In addition to antigen recognition, a co-stimulatory signal resulting from the B7-CD28 interaction is also necessary for activation. T-cell activation results in upregulation of PD1 on the cell surface and the secretion of proinflammatory cytokines, such as interferon gamma (IFNγ), into the tumor microenvironment. IFNγ induces the upregulation of PDL1 on DCs and tumor cells. The PD1/PDL1 interaction inhibits TCR signaling and leads to an increase in T regulatory (Treg) cells, which inhibit CD8+ T-cell activity and prevent clonal expansion. This adaptive immune resistance allows the tumor cells to evade a T-cell-mediated antitumor response.
However, induction of the PD1/PDL1 pathway has recently been shown in a number of adult malignancies as a means by which tumors suppress the host immune response. PDL1 expression has previously been documented in a number of adult tumors, including melanoma, renal, esophageal, breast, and lung carcinomas (26,32,33,34,35,36). Early animal data showed that tumor expression of PDL1 would inhibit the activity of infiltrating tumor-reactive T cells (26,37,38,39), and further studies demonstrated a correlation between expression of PDL1 and adverse outcomes in adult malignancies (40,41,42). Unfortunately, data regarding expression of PDL1 in pediatric cancers is minimal, and what does exist is derived from either animal models or study samples involving predominantly adult patient samples, with only a modest proportion of pediatric primary tumor samples. To date, there exist no published studies regarding expression patterns of PDL1 in Ewing sarcoma or rhabdomyosarcoma. Soft tissue sarcomas have been shown to express PDL1 (with corresponding expression of PD1 on tumor infiltrating lymphocytes), and this expression has been correlated with reduced survival (42). In a recent study, subsets of both adult and pediatric osteosarcoma samples were shown to express high levels of PDL1, with high levels of expression trending with decreased overall survival, although it did not quite achieve statistical significance (43). Another recent study demonstrated that PDL1 (along with PD1-positive CD8+ T cells) was upregulated in metastatic but not primary osteosarcoma, and inhibition of PD1 in a mouse model was able to decrease tumor burden and improve survival (44). PDL1 has also been shown to be expressed in adult samples of various hematologic malignancies, including acute myeloid leukemia, acute lymphoblastic leukemia, anaplastic large cell lymphoma, Hodgkin lymphoma, and diffuse large B-cell lymphoma (45,46,47,48,49).
Novel therapeutic agents that inhibit the PD1/PDL1 axis have shown promise in adult malignancies. Monoclonal antibodies (mAbs) against PDL1 and PD1 alike have produced therapeutic responses in significant subsets of patients with melanoma, renal, and lung cancer (50,51,52,53). Pembrolizumab (Keytruda, Merck, Kenilworth, NJ), formerly lambrolizumab (MK3475), became the first FDA-approved anti-PD1 mAb in September 2014 based on tumor responses and durability of the responses seen in early clinical trials. It is approved for use in patients with advanced melanoma who have progressed on ipilimumab and BRAF-directed therapy in patients who carry a BRAF mutation (54,55). Nivolumab (BMS-936558, Bristol-Meyers-Squibb, New York, NY), a fully humanized IgG4 anti-PD1 mAb, was initially shown in phase I clinical trials to induce objective responses in 18–28% of patients with melanoma, renal cell carcinoma (RCC), and non–small-cell lung carcinoma (NSCLC) (51). Subsequent phase II trials in metastatic RCC recapitulated objective responses in a subset (22–26%) of patients, as well as dose-dependent increases in overall survival (56). It is now FDA approved as monotherapy for treatment of unresectable or metastatic melanoma, and disease progression following treatment with ipilimumab and BRAF-directed therapy in patients with tumors possessing the BRAF mutation (57,58). Additionally, nivolumab has recently been FDA approved for combination therapy against NSCLC which has progressed after platinum-based chemotherapy, following the results of a recent phase III clinical trial which demonstrated superior osteosarcoma compared to standard of care (59). Pidilizumab (CT-011, CureTech, Yavne, Israel) is an anti-PD1 mAb which has been studied in phase I trials in a small sample of patients with diffuse large B-cell lymphoma, with 33% of patients demonstrating increased overall survival (60). A recent phase II clinical trial in which pidilizumab was administered to diffuse large B-cell lymphoma patients following autologous stem cell transplant showed increased duration of progression-free survival (61). Additionally, in studies involving nivolumab, pembrolizumab, and pidilizumab, many of the responders continued to demonstrate sustained regression even after the treatment was discontinued (50,51,52,53,54,55,56,57,58,59,60,61). Anti-PDL1 mAbs have shown similar efficacy in phase I clinical trials as well. One such mAb, BMS-936559, showed objective response rates in a small subset (10–17%) of melanoma, RCC, ovarian cancer, and NSCLC patients, with sustainable, durable tumor regression for over 1 year in nearly half of the responders (52). MPDL3280A, another anti-PDL1 mAb, induced objective responses in roughly 25% of melanoma, NSCLC, RCC, and metastatic bladder cancer patients (62,63). Multiple phase III clinical trials are now either recruiting or ongoing involving MPDL3280A, and it was recently granted breakthrough status by the FDA for treatment of NSCLC. Furthermore, clinical trials are currently being planned exploring the use of nivolumab, pembrolizumab, and pidilizumab in pediatric tumors. While many of these trials are not yet recruiting and therefore have yet to demonstrate therapeutic efficacy, they plan to explore the use of these agents against osteosarcoma, Ewing sarcoma, RMS, malignant gliomas, and soft tissue sarcoma ( Table 2 ).
While PD1/PDL1 blockade provides durable therapeutic responses in a subset of patients (many of whom have proven refractory to conventional chemotherapy), the response rate to therapy is still present in only a minority of patients. Therefore, identification of factors that predict a positive response to anti-PD1/PDL1 therapy is a rapidly growing area of interest. While early studies involving patients treated with a nivolumab showed that PDL1 expression in tumor tissues had the strongest association with therapeutic response to treatment (64), PDL1 expression is a dynamic phenomenon which is regulated by inflammatory cytokines within the tumor microenvironment (65,66). This induction of PDL1 via localized inflammation, termed adaptive immune resistance, requires the initial presence of tumor-infiltrating CD8+ T cells (26,39). These T cells recognize tumor antigens via TCRs, which triggers their initial activation, expression of PD1, and the production of local interferons as part of their initial antitumor response. These interferons then induce the expression of PDL1 in tumor cells, leading to the suppression of this immune response (67,68).
Recent work has demonstrated that tumor-infiltrating CD8+ T cells do indeed appear to be a prerequisite for an effective response to PD1/PDL1 inhibition. Tumeh et al. (69) recently showed that metastatic melanoma patients who responded to PD1 blockade with pembrolizumab had higher pretreatment levels of tumor-infiltrating CD8+ T cells expressing PD1, as well as higher PDL1 expression in the tumors themselves. Additionally, they were able to demonstrate that these T cells, in addition to functionally producing interferon, had a more clonal TCR population, implying specific targeting of the tumor itself. The fact that these clonal restrictions on tumor-infiltrating T cells, as well as the PD1/PDL1 expression patterns were present in the responder patients even prior to treatment with pembrolizumab, demonstrates the necessity for these preexisting immunologic conditions prior to successful therapeutic inhibition of PD1/PDL1. Additionally, the patients who responded to treatment were found to have further posttreatment proliferation of clonally expanded, TCR-restricted CD8+ T cells, and this proliferation was noted to correlate with reduction in tumor size.
This evidence of the need for a preexisting antitumor T-cell response for optimal effectiveness of PD1/PDL1 inhibition provides the pediatric oncology community with an opportunity for augmenting the potential effectiveness of existing experimental tumor vaccines via simultaneous inhibition of PD1/PDL1. While study of PDL1 expression patterns in pediatric tumors and clinical trials involving PD1/PDL1 inhibition is a logical extension of the highly promising work from adult cancers, synergizing techniques for the treatment of pediatric tumors could improve response rates. Indeed, pediatric tumors which are more immunogenic and possess inherent tumor-infiltrating T cells, such as osteosarcoma, may be the best candidates for PD1/PDL1 inhibition. This may be due to the fact that osteosarcomas are genetically diverse malignancies with multiple chromosomal abnormalities and hyperdiploidy (70), which may lend itself to greater antigenic variety increasing the likelihood of a T-cell response. Early attempts at immunotherapy against osteosarcoma (using interleukin and interferon administration to treat both local and metastatic disease, as well as granulocyte-macrophage colony-stimulating factor in metastatic disease) were based on this principle of augmenting existing immune responses to improve patient outcomes (16). Recent studies demonstrating the importance of CD8+ T-cell responses in better patient outcomes (71,72) have lead to additional immunotherapies which focus on selective augmentation of the T-cell response through other mechanisms, including selective expansion of T cells present in the tumor microenvironment, and even genetically engineered T cells with chimeric antigen receptors specific for a given tumor (13). While focused study of less genetically and immunogenically diverse, more primitive tumors such as Ewing’s sarcoma and rhabdomyosarcoma may demonstrate less robust PDL1 expression and PD1-positive CD8+ T-cell infiltration of the tumor, inducing such activity with either dendritic cell (DC) or T-cell-based tumor vaccines and subsequently combining PD1/PDL1 inhibition may yield superior clinical responses.
Combining PD1/PDL1 inhibition with DC-based vaccines that augment T-cell response to increase the likelihood of initial antitumor activity is already in the early stages of being studied. A recent study by Fu et al. (73) showed that an interferon-inducing cancer vaccine did indeed upregulate PDL1 expression in tumor tissue, as well as increasing the amount of tumor-infiltrating CD8+ T-cells. Moreover, coadministration of a PD1-blocking mAb caused a complete regression of established tumors in a mouse model of melanoma. Several other preclinical mouse models combining various tumor vaccines with anti-PD1/PDL1 antibodies have also demonstrated similarly enhanced CD8+ T-cell activity against a variety of solid tumors, including mouse models of sarcoma, pancreatic ductal adenocarcinoma, ovarian, breast, colon, and hepatocellular carcinoma (74,75,76,77,78). Given the wide variety of subtypes of tumor vaccines used in these studies (DNA vaccines, DC vaccines, peptide vaccines), it may be that any or all of them may prove useful for potential clinical trials.
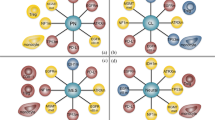
Additionally, PD1/PDL1 interactions between DCs and T cells lead to increases in Tregs, as well as concomitant decreases in CD8+ T-cell activity. Inhibition of DC-based PD1/PDL1 interactions in this context would itself directly contribute to the decreased Treg activity and enhanced CD8 clonal expansion needed for antitumor activity (79,80,81). Given how the majority of experimental pediatric sarcoma vaccines rely on tumor-pulsed DCs, we have the opportunity to enhance the potential effectiveness of these agents from multiple angles: triggering an inflammatory tumor microenvironment in which the DC primes and activates CD8+ T cells against a tumor, inhibiting the PD1/PDL1 interaction between the tumor and the CD8+ T cells to perpetuate their antitumor activity, and inhibiting the PD1/PDL1 interaction between the DCs and T cells to decrease Treg activity and enhance the clonal expansion of these tumor-specific CD8+ T cells ( Figure 2 ). Preclinical studies recapitulating the successes of adult cancers are necessary prior to the use of tumor vaccines in combination with PD1/PDL1 inhibitors in clinical trials for pediatric malignancies, and there may be challenges in making such therapeutics widely available and equally or more cost-effective than conventional medical therapy (for example, many tumor vaccines require individual tumor samples pulsed with harvested DCs, which would only be available at specialized centers). However, if such studies are successful, they could yield new treatment options for aggressive pediatric tumors and potentially improve their otherwise bleak prognosis.

Tumor-pulsed DCs activate CD8+ T cells against tumor-specific antigens and increase the number of CD8+ T cells. Inhibition of the PD1/PDL1 interaction between T cells and tumor cells through the use of monoclonal antibodies increases cytotoxic T-cell antitumor response. Furthermore, blockade of this interaction between T cells and DCs reduces Treg activity and enhances CD8+ T-cell clonal expansion. Since response to PD1/PDL1 blockade requires the presence of CD8+ T cells, vaccine-induced DCs can be utilized to increase the number of CD8+ T cells and maximize antitumor activity.
Conclusions
The value of anti-PD1/PDL1 therapy in cancer has been shown in multiple adult malignancies, and the further study of this system and its therapeutic inhibition in pediatric sarcomas and other pediatric solid tumors is a logical extension of this body of work. Providing additional opportunity, however, is the mounting evidence that a preexisting immune response to tumors is both necessary and sufficient for a therapeutic response to PD1/PDL1 blockade. The growing number of preclinical trials demonstrating that PD1/PDL1 inhibition, combined with cancer vaccines which generate directed T-cell responses against tumors, greatly enhances tumor regression and clinical responses provides a means to further utilize existing pediatric cancer vaccines to enhance their effectiveness. By instigating a directed T-cell immune response against pediatric malignancies with personalized cancer vaccines, and by then sustaining that response via inhibition of the adaptive immune resistance of PDL1 induction caused by this T-cell response, we have the potential to create novel, personalized therapeutic options and improve the clinical outcomes of some of the most aggressive and deadly pediatric tumors.
Statement of Financial Support
No financial assistance was received to support this study.
Disclosure
The authors have no financial conflicts of interest to disclose.
References
Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 2014;64:83–103.
Musselman JR, Spector LG, Krailo MD, et al. The Children’s Oncology Group Childhood Cancer Research Network (CCRN): case catchment in the United States. Cancer 2014;120:3007–15.
Smith MA, Altekruse SF, Adamson PC, Reaman GH, Seibel NL. Declining childhood and adolescent cancer mortality. Cancer 2014;120:2497–506.
Breneman JC, Lyden E, Pappo AS, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma–a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol 2003;21:78–84.
Pappo AS, Anderson JR, Crist WM, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol 1999;17:3487–93.
Stahl M, Ranft A, Paulussen M, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 2011;57:549–53.
Hawkins DS, Arndt CA. Pattern of disease recurrence and prognostic factors in patients with osteosarcoma treated with contemporary chemotherapy. Cancer 2003;98:2447–56.
Bielack SS, Kempf-Bielack B, Branscheid D, et al. Second and subsequent recurrences of osteosarcoma: presentation, treatment, and outcomes of 249 consecutive cooperative osteosarcoma study group patients. J Clin Oncol 2009;27:557–65.
Oeffinger KC, Mertens AC, Sklar CA, et al.; Childhood Cancer Survivor Study. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 2006;355:1572–82.
Spunt SL, Smith LM, Ruymann FB, et al. Cyclophosphamide dose intensification during induction therapy for intermediate-risk pediatric rhabdomyosarcoma is feasible but does not improve outcome: a report from the soft tissue sarcoma committee of the children’s oncology group. Clin Cancer Res 2004;10:6072–9.
Granowetter L, Womer R, Devidas M, et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children’s Oncology Group Study. J Clin Oncol 2009;27:2536–41.
Lewis IJ, Nooij MA, Whelan J, et al.; MRC BO06 and EORTC 80931 collaborators; European Osteosarcoma Intergroup. Improvement in histologic response but not survival in osteosarcoma patients treated with intensified chemotherapy: a randomized phase III trial of the European Osteosarcoma Intergroup. J Natl Cancer Inst 2007;99:112–28.
Mackall CL, Merchant MS, Fry TJ. Immune-based therapies for childhood cancer. Nat Rev Clin Oncol 2014;11:693–703.
Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998;392:245–52.
Ribas A, Butterfield LH, Glaspy JA, Economou JS. Current developments in cancer vaccines and cellular immunotherapy. J Clin Oncol 2003;21:2415–32.
Wilky BA, Goldberg JM. Immunotherapy in sarcoma: a new frontier. Discov Med 2014;17:201–6.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004;10:909–15.
Gong J, Chen D, Kashiwaba M, Kufe D. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nat Med 1997;3:558–61.
Gong J, Nikrui N, Chen D, et al. Fusions of human ovarian carcinoma cells with autologous or allogeneic dendritic cells induce antitumor immunity. J Immunol 2000;165:1705–11.
Gong J, Chen D, Kashiwaba M, et al. Reversal of tolerance to human MUC1 antigen in MUC1 transgenic mice immunized with fusions of dendritic and carcinoma cells. Proc Natl Acad Sci USA 1998;95:6279–83.
Finke LH, Wentworth K, Blumenstein B, Rudolph NS, Levitsky H, Hoos A. Lessons from randomized phase III studies with active cancer immunotherapies–outcomes from the 2006 meeting of the Cancer Vaccine Consortium (CVC). Vaccine 2007;25:Suppl 2:B97–B109.
Finkelstein SE, Iclozan C, Bui MM, et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys 2012;82:924–32.
Madan RA, Gulley JL, Fojo T, Dahut WL. Therapeutic cancer vaccines in prostate cancer: the paradox of improved survival without changes in time to progression. Oncologist 2010;15:969–75.
Stein WD, Gulley JL, Schlom J, et al. Tumor regression and growth rates determined in five intramural NCI prostate cancer trials: the growth rate constant as an indicator of therapeutic efficacy. Clin Cancer Res 2011;17:907–17.
Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010;28:4722–9.
Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med 2012;366:2517–9.
Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 2006;203:883–95.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010;236:219–42.
Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
Fife BT, Pauken KE, Eagar TN, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol 2009;10:1185–92.
Amarnath S, Mangus CW, Wang JC, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med 2011;3:111ra120.
Atefi M, Avramis E, Lassen A, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res 2014;20:3446–57.
Jilaveanu LB, Shuch B, Zito CR, et al. PD-L1 expression in clear cell renal cell carcinoma: an analysis of nephrectomy and sites of metastases. J Cancer 2014;5:166–72.
Ohigashi Y, Sho M, Yamada Y, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res 2005;11:2947–53.
Velcheti V, Schalper KA, Carvajal DE, et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest 2014;94:107–16.
Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 2007;19:813–24.
Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8:793–800.
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA 2002;99:12293–7.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–64.
Hamanishi I, Mandal M, Iwasaki M, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA 2007;104:3360–3365.
Thompson RH, Gillett MD, Cheville JC, et al. Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA 2004;101:17174–9.
Kim JR, Moon YJ, Kwon KS, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS One 2013;8:e82870.
Shen JK, Cote GM, Choy E, et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol Res 2014;2:690–8.
Lussier DM, O’Neill L, Nieves LM, et al. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J Immunother 2015;38:96–106.
van Dam LS, de Zwart VM, Meyer-Wentrup FA. The role of programmed cell death-1 (PD-1) and its ligands in pediatric cancer. Pediatr Blood Cancer 2015;62:190–197.
Chen X, Liu S, Wang L, Zhang W, Ji Y, Ma X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol Ther 2008;7:622–7.
Afreen S, Dermime S. The immunoinhibitory B7-H1 molecule as a potential target in cancer: killing many birds with one stone. Hematol Oncol Stem Cell Ther 2014;7:1–17.
Andorsky DJ, Yamada RE, Said J, Pinkus GS, Betting DJ, Timmerman JM. Programmed death ligand 1 is expressed by non-hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin Cancer Res 2011;17:4232–44.
Yamamoto R, Nishikori M, Tashima M, et al. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci 2009;100:2093–100.
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med 2014;65:185–202.
Dummer R, Daud A, Puzanov I, et al. A randomized controlled comparison of pembrolizumab and chemotherapy in patients with ipilimumab-refractory melanoma. J Transl Med. 2015;13:2062.
Robert C, Schachter J, Long GV, et al.; KEYNOTE-006 investigators. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol 2015;33:1430–7.
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
Sundar R, Cho BC, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol 2015;7:85–96.
Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res 2008;14:3044–51.
Armand P, Nagler A, Weller EA, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol 2013;31:4199–206.
Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014;515:558–62.
Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064–74.
Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013;5:200ra116.
Bald T, Landsberg J, Lopez-Ramos D, et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov 2014;4:674–87.
Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4:127ra37.
Ribas A, Tumeh PC. The future of cancer therapy: selecting patients likely to respond to PD1/L1 blockade. Clin Cancer Res 2014;20:4982–4.
Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
Martin JW, Squire JA, Zielenska M. The genetics of osteosarcoma. Sarcoma 2012;2012:627254.
Fritzsching B, Fellenberg J, Moskovszky L, et al. CD8(+)/FOXP3(+)-ratio in osteosarcoma microenvironment separates survivors from non-survivors: a multicenter validated retrospective study. Oncoimmunology 2015;4:e990800.
Théoleyre S, Mori K, Cherrier B, Passuti N, Gouin F, Rédini F, Heymann D. Phenotypic and functional analysis of lymphocytes infiltrating osteolytic tumors: use as a possible therapeutic approach of osteosarcoma. BMC Cancer 2005;5:123.
Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res 2014;74:4042–52.
Soares KC, Rucki AA, Wu AA, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother 2015;38:1–11.
Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 2013;73:6900–12.
Karyampudi L, Lamichhane P, Scheid AD, et al. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res 2014;74:2974–85.
Sawada Y, Yoshikawa T, Shimomura M, Iwama T, Endo I, Nakatsura T. Programmed death-1 blockade enhances the antitumor effects of peptide vaccine-induced peptide-specific cytotoxic T lymphocytes. Int J Oncol 2015;46:28–36.
Rekoske BT, Smith HA, Olson BM, Maricque BB, McNeel DG. PD-1 or PD-L1 blockade restores antitumor efficacy following SSX2 epitope-modified DNA vaccine immunization. Cancer Immunol Res 2015;3:946–55.
Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol 2003;170:1257–66.
Curiel TJ, Wei S, Dong H, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med 2003;9:562–7.
Rosenblatt J, Glotzbecker B, Mills H, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother 2011;34:409–18.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Nowicki, T., Anderson, J. & Federman, N. Prospective immunotherapies in childhood sarcomas: PD1/PDL1 blockade in combination with tumor vaccines. Pediatr Res 79, 371–377 (2016). https://doi.org/10.1038/pr.2015.246
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.246
