
Abstract
We performed a genetic screen in mice to identify candidate genes that are associated with leukaemogenesis in the context of Trp53 heterozygosity. To do this we generated Trp53 heterozygous mice carrying the T2/Onc transposon and SB11 transposase alleles to allow transposon-mediated insertional mutagenesis to occur. From the resulting leukaemias/lymphomas that developed in these mice, we identified nine loci that are potentially associated with tumour formation in the context of Trp53 heterozygosity, including AB041803 and the Jun dimerization protein 2 (Jdp2). We show that Jdp2 transcriptionally regulates the Trp53 promoter, via an atypical AP-1 site, and that Jdp2 expression negatively regulates Trp53 expression levels. This study is the first to identify a genetic mechanism for tumour formation in the context of Trp53 heterozygosity.
Similar content being viewed by others


Introduction
Genetic alterations of TP53 are frequent events in tumourigenesis and promote genomic instability, impair apoptosis, and contribute to aberrant self-renewal.1, 2, 3, 4 The spectrum of mutations that occur in TP53 in human cancers is diverse. Missense mutations that deregulate the DNA-binding domain are common, and prevent or impair the transcriptional regulatory activity of TP53.3 Cytogenetic alterations that delete or disrupt TP53 have also been reported, as has epigenetic silencing due to methylation of the TP53 promoter.3 In both carcinomas and haematopoietic malignancies, TP53 mutation status has been shown to correlate with prognosis.5, 6 TP53 is generally considered a tumour-suppressor gene,2 with inactivation of both copies of the gene seen in many tumours. Paradoxically, several studies have observed accelerated tumourigenesis in Trp53+/− mice that develop tumours despite retaining a wild-type copy of Trp53.7, 8, 9, 10 Furthermore the analysis of tumours from Li-Fraumeni patients with germline alterations of TP53, suggest that a significant proportion may retain a wild-type TP53 allele.11, 12, 13 Threshold levels of P53 are required for processes such as suppression of apoptosis or induction of cell-cycle arrest.14 In the context of TP53 heterozygosity it is possible that transcriptional silencing of the wild-type TP53 allele by mechanisms such as promoter methylation, altered cis-regulation of the gene that decreases transcription from the wild-type TP53 allele, or post-translational modification of P53, decreases TP53 function to a level such that tumourigenesis can occur. In this paper we set out to identify, which somatically mutated genes can contribute to tumour formation in the context of Trp53 heterozygosity. To do this we used mice heterozygous for Trp53 (as well as Trp53 wild type and null controls) to genetically dissect this phenomenon focusing on leukaemogenesis as a model system. This analysis allowed us to identify nine loci that are potentially associated with tumour formation in the context of Trp53 heterozygosity. We show that the Jun dimerization protein 2 (Jdp2) is a site frequently targeted by transposon insertion events leading to upregulated Jdp2 expression and a decrease in Trp53 expression levels. Further we illustrate that Jdp2 regulates the Trp53 promoter via an atypical AP-1 binding site. This study is the first to identify a genetic mechanism for tumour formation in the context of Trp53 heterozygosity.
Results and discussion
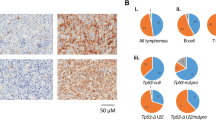
Mouse lines carrying the mutant Trp53 allele, Trp53Tyr, which are null for Trp53,15 the SB (Sleeping Beauty) transposon array, T2/Onc,16 and the SB transposase allele, Rosa26SB11 (see Dupuy et al.17) were intercrossed to generate mice that were homozygous, heterozygous or wild type for the Trp53Tyr allele (hereafter referred to as Trp53−/−, Trp53+/− or Trp53+/+ mice, respectively) with or without SB transposition occurring (that is, on a T2/Onc+/Tg Rosa26+/SB11 or T2/Onc+/+, Rosa26+/SB11 background, respectively). These mice were aged until they became moribund, and, as expected, SB transposition significantly accelerated tumour latency in mice of all genotypes (Figure 1a). The predominant tumour type of all genotypes was a widely disseminated CD3+ T-cell lymphoma (Figures 1b and c). A number of solid tumours, mainly undifferentiated sarcomas, were also observed, but only in Trp53+/− or Trp53−/− mice (Figures 1b and c).

Loss of Trp53 promotes tumourigenesis. (a) Kaplan–Meier curves showing the tumour latency in Trp53+/+, Trp53+/− and Trp53−/− mice on a transposon ‘jumping’ background (that is, T2/Onc+/Tg Rosa+/SB11 solid lines) and ‘non-jumping’ control background (that is, T2Onc+/+; Rosa26+/SB11 dashed lines). Curve comparison using the log-rank (Mantel–Cox) test: P<0.0001 for Trp53+/+ vs Trp53+/− vs Trp53−/− mice on a ‘jumping’ background. (b) Categorisation of the malignancies developed by the Trp53+/+, Trp53+/− and Trp53−/− mice on a ‘jumping’ background according to the tumour type. Several mice had multiple tumour types. Numbers in brackets represent the percentage of mice developing a specific tumour type as a proportion of the genotype. (c) Representative photomicrographs of formalin-fixed, hematoxylin- and eosin-stained sections of (i) thymic lymphoma, (ii) splenic lymphoma, (iii) undifferentiated sarcoma and (iv) osteosarcoma. Representative photomicrographs of immunohistochemically-stained liver sections infiltrated by lymphomas of (v) B-cell origin (CD45R+) or (vi) T-cell origin (CD3+). Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections that had undergone antigen retrieval (microwaving in high pH citrate buffer for 3 × 5 min) using rabbit anti-human polyclonal CD3 antibody (Dako, Ely, UK) and rat anti-mouse/human monoclonal B220/CD45R antibody (BD Biosciences, Oxford, UK). The immunohistochemical signal was detected using a secondary biotinylated goat anti-rabbit or anti-rat antibody (Vector Laboratories, Burlingame, CA, USA), using the Vectorstain Elite ABC kit (Vector Laboratories) according to the manufacturer's instructions. All sections shown are representative and images are at × 400 magnification.
Genomic DNA from 36 Trp53+/+, 116 Trp53+/− and 9 Trp53−/− SB-induced leukaemic/lymphomic tissues (typically spleen, thymus or lymph node) was extracted and subjected to a previously described linker-mediated PCR approach18 to amplify barcoded genomic fragments containing transposon–genome junction sequences. These products were then pooled and sequenced on the 454 platform, from which we generated 487 586 uniquely mapped sequence reads (approximately 3000 per tumour). After merging overlapping reads originating from the same sample and removing any on chromosome 1 (because SB transposons frequently reintegrate into regions adjacent to the donor locus—a phenomenon known as ‘local hopping’16), we obtained 7538 (Trp53+/+), 21 975 (Trp53+/−) and 1829 (Trp53−/−) unique, non-redundant insertion sites (for the respective tumour genotypes indicated in brackets). Using a previously described Gaussian Kernel Convolution statistical method for determining common insertion sites (CISs),19, 20 we identified 42, 63 and 9 CISS in Trp53+/+, Trp53+/− and Trp53−/− tumours, respectively, (P<0.05 on a chromosome-adjusted scale; Figure 2 and Supplementary Table 1). Many of these genes have been previously implicated in the pathogenesis of T-cell lymphomagenesis/leukaemogenesis, including NOTCH1, PTEN and IKZF1 (reviewed in Demarest et al.21). There were 12 CIS genes in common between the Trp53+/+ and Trp53+/− tumours, specifically Mecom (Mds1 and Evi1 complex locus), Myb, Notch1, Stat5b, Erg, Ikzf1, Raf1, Rasgrp1, Zmiz1, Pten, AB041803 and Il2rb. Given that eight of these genes have also been identified as CISS in leukaemias/lymphomas from T2/Onc;Rosa26-SB11 mice on a wild-type background (Myb, Notch1, Erg, Ikzf1, Rasgrp1, Zmiz1, Pten and AB041803),22 they likely represent genes involved in lymphomagenesis/leukemogenesis in general, and do not contribute to promotion of tumourigenesis in the context of Trp53 heterozygosity.

Analysis of common insertion sites. A Circos plot showing the common insertion sites (CISS) called in tumours from Trp53+/+ (black), Trp53+/− (blue) and Trp53−/− (red) mice. Lines crossing the circle indicate statistically significant co-occurring mutations with the thickness of the line indicating the level of significance. All tumour DNA was extracted using GenePure kits (Qiagen, Sussex, UK) and transposon insertion site sequences were generated on the 454 platform (Roche, West Sussex, UK), as described previously.30 Processing of 454 reads, identification of insertion sites, and the Gaussian Kernel Convolution statistical methods used to identify CISS have been described previously.19, 20 The P-value for each CIS was calculated using an adjusted-by-genome cutoff of P<0.05. A complete list of the CISS is given in Supplementary Table 1.
A CIS gene that was found in the Trp53+/− and Trp53−/− tumours, but not Trp53+/+ tumours, was Rapgef6. The Rap1 guanine nucleotide exchange factor RAPGEF6 (also known as PDZGEF2) has a critical role in the maturation of adherens junctions.23 Although no immediate role for RAPGEF6 in tumourigenesis is evident, it has been shown to form protein complexes that result in the activation of Rap1A and control of cell adhesion/migration.24, 25 Interestingly, apart from Rapgef6, the CIS genes found in the Trp53−/− tumours were not found in tumours of the other genotypes. These included genes Usp42 (ubiquitin specific peptidase 42) and Wdr33 (WD repeat-containing protein 33). Although little is known about Wdr33 gene, Usp42 gene has been recently identified as a fusion partner of RUNX1 in three cases of myeloid neoplasia, and the associated upregulated expression of USP42 suggests a role of this deubiquitinating enzyme in the pathogenesis of this leukaemia.26
There were also four CISS that were found to co-occur in tumours (Figure 2), specifically Notch1 and Pten, Notch1 and Ikzf1, Pten and Ikzf1 and Pten and Akt2 in Trp53+/− tumours. These genes have all been previously implicated in the pathogenesis of T-ALL.21, 27 Our results are in keeping with the literature, as there is evidence for genetic co-operation of these genes in development of T-ALL. For example, loss of Ikzf1, a direct repressor of Notch target genes, and suppression of p53-mediated apoptosis are essential for development of T-ALL and PTEN inactivation can compensate for some Notch-mediated processes in T-ALL.21 In addition, retroviral insertional mutagenesis recently identified Ikzf1, KrasG12D and Notch1 as a novel genetic pathway in T-lineage leukaemogenesis.28
Quantitative PCR was performed on all tumours from Trp53+/− mice to identify those that had retained a wild-type copy of Trp53 and those that carried two copies of the targeted Trp53Tyr allele (presumably having lost the wild-type allele by mitotic recombination; Figure 3a). From the 111 Trp53+/− tumours analysed, we identified 40 that had retained a wild-type Trp53 allele (defined as having a normalised wild-type allele content of >0.28 and a Trp53Tyr allele content of <0.7) and 27 tumours that carried two targeted Trp53Tyr alleles and no wild-type allele signal (defined as having a normalised wild-type allele content of <0.1 and a Trp53Tyr allele content of >0.8). To determine if there were any somatic mutations in the intact wild-type copy of Trp53, genomic DNA from all 111 Trp53+/− tumours (as well as some tail samples to facilitate the identification of somatic mutations) underwent Trp53 sequencing on the Illumina platform (Illumina, San Diego, CA, USA) to scan for point mutations (using the primers shown in Supplementary Table 2). Paired-end sequencing of PCR amplified fragments was followed by base-calling with SAMTOOLS mpileup,29 which identified three possible mutations, specifically MMU11:69400422 (T-C), MMU11:69403089 (G-A) and MMU11:69403110 (G-A) in single tumours. All other tumours appeared to have retained the wild-type Trp53 allele. A further two sequence changes at MMU11:69401065 and MMU11:69401996 were discovered in 22 and 44 of the samples, respectively, and are therefore likely to be germline variants (as these mice were on a mixed C57BL/6J-129Sv background and the sequencing data was compared with the C57BL/6J reference genome). These data suggest point mutations of the wild-type Trp53 are infrequent in our model.

Identification of driver mutations associated with loss of Trp53 by mitotic recombination or with the retention of a wild-type copy of Trp53. (a) SYBR Green quantitative real-time PCR (ABI, Carlsbad, CA, USA) was performed on tumour genomic DNA to quantify the relative proportions of Trp53 wild-type and Trp53Tyr alleles in genomic DNA extracted from the leukaemias/lymphomas and data were normalised to the single-copy genes β-Actin and Gapdh (primers are detailed in Supplementary Table 3). Red triangles represent tumours from Trp53−/− mice, blue squares represent tumours from Trp53+/− mice and black circles represent tumours from Trp53+/+ mice. Of the tumours from Trp53+/− mice: open squares are those that have retained a wild-type copy of Trp53, closed dark blue squares are those that have lost the wild-type Trp53 allele by mitotic recombination (MR) and closed light blue squares are those with a mixture of Trp53+/− and Trp53−/− cells and thus were excluded from further analyses. (b). Common insertion sites (CISs) were identified in tumours from Trp53+/− mice that had retained a wild-type copy of Trp53 (dotted blue circle) and those that had lost the wild-type copy (solid blue circle) as described previously.19, 20 CISS were called using a genome wide cut-off of P<0.05. Asterisk indicates the CIS was also found in the other genotype/circle, but below the P<0.05 cut-off. Double asterisk indicates the CISS were in intergenic regions (that is, not located within ±150K base pairs of a gene and were given the label ‘CIS’ followed by the chromosome and the peak location of the Gaussian kernel; there were two regions for ‘tumours retaining a wild-type copy of Trp53’: CIS7:37317163_15k and CIS5:75854217_15k, and one for ‘tumours without a wild-type copy of Trp53’: CIS7:37322632_15k) (c). Location and orientation of the transposon insertions (blue triangles) associated with the Jdp2 CIS (the exons of Jdp2 are represented as boxes). One tumour was found to harbour multiple independent transposon insertion events (indicated with dotted lines). (d). Quantitative PCR (qPCR) was performed on five tumours containing insertions in Jdp2 and nine randomly selected Trp53+/− T-cell tumours (without insertions in Jdp2). RNA was extracted using the RNeasy Minikit (Qiagen), DNAse-treated (Turbo DNase, Ambion, Warrington, UK) and reverse transcribed (RNA to cDNA EcoDry Random Hexamers, Clontech, Mountain View, CA, USA) according to the manufacturer's instructions. Quantitative PCR was performed in triplicate using SYBR Green PCR MasterMix (Applied Biosystems, Carlsbad, CA, USA) and the CT for Trp53 and Jdp2 were normalized to the ‘control’ (average of five housekeeping genes: Gapdh, β-Actin, Hprt1, Rpl32 and Rpl13a) using the 2–CT method.42 Primers used for qPCR are given in Supplementary Table 4. (e). Transient overexpression of JDP2 in NIH3T3 cells resulted in a significant repression of Trp53 proximal promoter activity. The 375 bp mouse Trp53 proximal promoter (Trp53-Luc) was PCR amplified from tail genomic DNA (using primers: F: 5′-AAAAAAAAGGTACCGGTCCACTTACGATAAAAAC-3′ and R: 5′-AAAAAAAAAAGATCTGGTCCCAATGAACTGAAGCT-3′) and cloned into the pGL3-BASIC vector (Promega, Southhampton, UK). The mutated mouse Trp53 proximal promoter in which the 7 bp PF-1 site (5′-TGACTCT-3′) was removed (mutTrp53-Luc) was synthesized (GeneArt-Invitrogen, Paisley, UK) and cloned into the pGL3-BASIC vector. A full-length human JDP2 cDNA was obtained from Origene (Rockville, MD, USA). NIH3T3 cells grown in 96-well plates were transfected with (i) either 100 ng Trp53-Luc (black lines) or mutTrp53-Luc (grey lines), (ii) 20 ng pRL-SV40 (an internal control reporter; Promega) and (iii) either 50 ng JDP2 cDNA or empty vector according to the manufacturer's instructions (Lipofectamine 2000; Invitrogen). Firefly and Renilla luciferase were measured 50 h later using the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions (Promega). The firefly light units were normalised to the Renilla light units. All data were normalised to the average value of the ‘control’ transfection (Luc vector plus empty vector) and were presented as fold-change relative to the control. Experiments were performed in triplicate on at least three independent occasions and the data analysed by two-tailed Student's t-test. (f). Transient overexpression of JDP2 in HEK293T cells represses TRP53 expression. HEK293T cells (Gryphon Eco, Allele Biotechnology, San Diego, CA, USA) were seeded in 12-well plates and transfected with 2 μg Myc-DDK-tagged ORF clone of JDP2 (pCMV6Entry; Origene) or empty vector, according to the manufacturers' instructions (Lipofectamine 2000, Invitrogen). Experiments were performed in triplicate. RNA was extracted 48 h post-transfection and reverse transcribed as described above. Quantitative PCR was performed in triplicate using SYBR Green PCR MasterMix (Applied Biosystems) and the CT for TP53 and JDP2 were normalized as described above. Primers used for qPCR are given in Supplementary Table 4.
Taking the insertion sites found in tumours from Trp53+/− mice, we performed CIS analysis in two ways. First, the tumours were divided into two groups: those that had either retained a wild-type copy of Trp53 or those that had lost the wild-type copy to identify the CISS that were unique and common to each group (Figure 3b). We found a set of nine CIS loci enriched in Trp53+/− mice that developed tumours despite retaining a wild-type copy of the gene, including AB041803, Akt2, Eras, Ikzf1, Jdp2, Myb, Rapgef6 and two intergenic regions. Second, we pooled the insertion sites from both groups together and then distinguished genotype-specific CISS using a P-value generated by Fisher's Exact test analysis.30 Using this more ‘stringent’ method of CIS calling, we identified two CISS that were ‘enriched’ in Trp53+/− tumours that had retained a wild-type copy of Trp53, specifically AB041803 and Jdp2. Little is known about AB041803 and as yet no role in tumourigenesis is evident. In addition, it was also found to be a CIS in leukaemia/lymphoma of wild-type mice (Supplementary Table 1).22 Thus we focused on Jdp2.
Transcription factor JDP2 (also known as JUNDM2) is an AP-1 repressor protein31 that has a paradoxical role in tumour formation. Overexpression of Jdp2 has been shown to potentiate hepatocellular carcinoma in mice32 and retroviral insertions predicted to activate the gene have been reported in mouse lymphoma models.33, 34 In contrast, downregulation of JDP2 has been associated with a poor prognosis in pancreatic cancer.35 Loss of Jdp2 has also been associated with resistance to replicative senescence,36, 37 and Jdp2 expression has been shown to suppress cell-cycle progression by downregulation of cyclin-A2.38 However, hypomethylation of the Jdp2 promoter or upregulation of Jdp2 expression in common myeloid progenitors and in granulocyte-macrophage progenitors has led to suggestions that it functions as a regulator of myelopoiesis.39 Here, we find that transposon insertions in the Jdp2 promoter occur exclusively in tumours from Trp53+/− mice that retain a wild-type allele of Trp53. These insertions clustered in the promoter of Jdp2 (Figure 3c) and were mostly orientated so that the transposon was inserted in the same transcriptional orientation as the gene, suggesting that these insertions were functioning to drive overexpression (with a single insertion orientated on the reverse strand relative to the gene, which may represent an enhancer insertion40). RT–PCR on RNA from these tumours showed splicing of the T2Onc transposon splice donor site directly onto Jdp2 exons 2 and/or 3 (Supplementary Figure 1). Indeed insertions in this exact location have been shown to activate Jdp2 expression,34 and consistent with this, qPCR on RNA from tumours containing insertions in Jdp2 showed a trend towards having higher expression levels of Jdp2 and lower expression levels of Trp53, relative to Trp53+/− tumours with no insertions in Jdp2 (randomly selected from mice on this study that had not lost the Trp53 allele by mitotic recombination; Figure 3d).
Co-transfection of JDP2 cDNA in an overexpression vector with a mouse Trp53 proximal promoter construct in murine NIH3T3 (Figure 3e) and human HEK293T cells (data not shown) resulted in significant repression of Trp53 promoter activity, confirming that overexpression of JDP2 functions directly on the Trp53 promoter to repress Trp53 expression. The ability of JDP2 to repress transcription of the p53 promoter is reported to occur via its binding to an atypical AP-1 site, termed the PF-1 site, in the p53 promoter.41 When we mutated (deleted) this binding site in the proximal Trp53 promoter, this completely abrogated the suppressive effects of JDP2 (Figure 3e), confirming that overexpression of JDP2 mediates repression of Trp53 through the PF-1 site in the proximal promoter. Furthermore, overexpression of JDP2 in HEK293T cells was shown to repress endogenous TP53 expression (Figure 3f).
In conclusion, we show that overexpression of Jdp2 in tumours that are heterozygous for Trp53 precludes the need for loss of the wild-type allele of Trp53 during the process of leukaemogenesis. Jdp2 overexpression is the first genetic mechanism that has been identified as being responsible for tumour formation in the context of Trp53 heterozygosity.
References
Levine AJ . p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331.
Giaccia AJ, Kastan MB . The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998; 12: 2973–2983.
Olivier M, Hollstein M, Hainaut P . TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2010; 2: a001008.
Goh AM, Coffill CR, Lane DP . The role of mutant p53 in human cancer. J Path 2011; 223: 116–126.
Robles AI, Harris CC . Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol 2010; 2: a001016.
Trbusek M, Smardova J, Malcikova J, Sebejova L, Dobes P, Svitakova M et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol 2011; 29: 2703–2708.
Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D et al. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J 1998; 17: 4657–4667.
Venkatachalam S, Tyner SD, Pickering CR, Boley S, Recio L, French JE et al. Is p53 haploinsufficient for tumor suppression? Implications for the p53+/- mouse model in carcinogenicity testing. Toxicol Pathol 2001; 29: 147–154.
Donehower LA . Using mice to examine p53 functions in cancer, aging, and longevity. Cold Spring Harb Perspect Biol 2009; 1: a001081.
Donehower LA, Lozano G . 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer 2009; 9: 831–841.
Li FP, Fraumeni Jr JF . Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 1969; 71: 747–752.
Malkin D, Li FP, Strong LC, Fraumeni Jr JF, Nelson CE, Kim DH et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250: 1233–1238.
Sedlacek Z, Kodet R, Kriz V, Seemanova E, Vodvarka P, Wilgenbus P et al. Two Li-Fraumeni syndrome families with novel germline p53 mutations: loss of the wild-type p53 allele in only 50% of tumours. Br J Cancer 1998; 77: 1034–1039.
Maiguel DA, Jones L, Chakravarty D, Yang C, Carrier F . Nucleophosmin sets a threshold for p53 response to UV radiation. Mol Cell Biol 2004; 24: 3703–3711.
Zheng B, Vogel H, Donehower LA, Bradley A . Visual genotyping of a coat color tagged p53 mutant mouse line. Cancer Biol Ther 2002; 1: 433–435.
Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA . Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 2005; 436: 272–276.
Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA . Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 2005; 436: 221–226.
Uren AG, Mikkers H, Kool J, van der Weyden L, Lund AH, Wilson CH et al. A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat Protoc 2009; 4: 789–798.
de Ridder J, Uren A, Kool J, Reinders M, Wessels L . Detecting statistically significant common insertion sites in retroviral insertional mutagenesis screens. PLoS Comput Biol 2006; 2: e166.
March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Eldridge M et al. Insertional mutagenesis reveals multiple networks of co-operating genes driving intestinal mutagenesis. Nat Genet 2011; 43: 1202–1209.
Demarest RM, Ratti F, Capobianco AJ . It's T-ALL about Notch. Oncogene 2008; 27: 5082–5091.
Collier LS, Adams DJ, Hackett CS, Bendzick LE, Akagi K, Davies MN et al. Whole-body sleeping beauty mutagenesis can cause penetrant leukemia/lymphoma and rare high-grade glioma without associated embryonic lethality. Cancer Res 2009; 69: 8429–8437.
Dubé N, Kooistra MR, Pannekoek WJ, Vliem MJ, Oorschot V, Klumperman J et al. The RapGEF PDZ-GEF2 is required for maturation of cell-cell junctions. Cell Signal 2008; 20: 1608–1615.
Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA . Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell 2009; 20: 1916–1925.
Iwasaki M, Tanaka R, Hishiya A, Homma S, Reed JC, Takayama S . BAG3 directly associates with guanine nucleotide exchange factor of Rap1, PDZGEF2, and regulates cell adhesion. Biochem Biophys Res Commun 2010; 400: 413–418.
Giguère A, Hébert J . Microhomologies and topoisomerase II consensus sequences identified near the breakpoint junctions of the recurrent t(7;21)(p22;q22) translocation in acute myeloid leukemia. Genes Chromosomes Cancer 2011; 50: 228–238.
Gutierrez A, Grebliunaite R, Feng H, Kozakewich E, Zhu S, Guo F et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J Exp Med 2011; 208: 1595–1603.
Dail M, Li Q, McDaniel A, Wong J, Akagi K, Huang B et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci USA 2010; 107: 5106–5111.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M et al. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell 2008; 133: 727–741.
Aronheim A, Zandi E, Hennemann H, Elledge SJ, Karin M . Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol Cell Biol 1997; 17: 3094–3102.
Bitton-Worms K, Pikarsky E, Aronheim A . The AP-1 repressor protein, JDP2, potentiates hepatocellular carcinoma in mice. Mol Cancer 2010; 9: 54.
Stewart M, Mackay N, Hanlon L, Blyth K, Scobie L, Cameron E et al. Insertional mutagenesis reveals progression genes and checkpoints in MYC/Runx2 lymphomas. Cancer Res 2007; 67: 5126–5133.
Rasmussen MH, Wang B, Wabl M, Nielsen AL, Pedersen FS . Activation of alternative Jdp2 promoters and functional protein isoforms in T-cell lymphomas by retroviral insertion mutagenesis. Nuc Acids Res 2009; 37: 4657–4671.
Yuanhong X, Feng X, Qingchang L, Jianpeng F, Zhe L, Kejian G . Downregulation of AP-1 repressor JDP2 is associated with tumor metastasis and poor prognosis in patients with pancreatic carcinoma. Int J Biol Markers 2010; 25: 136–140.
Nakade K, Pan J, Yamasaki T, Murata T, Wasylyk B, Yokoyama KK . JDP2 (Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative senescence. J Biol Chem 2009; 284: 10808–10817.
Wang SW, Lee JK, Ku CC, Chiou SS, Ho MF, Wu DC et al. Jun dimerization protein 2 in oxygen restriction; control of senescence. Curr Pharm Des 2011; 17: 2278–2289.
Pan J, Nakade K, Huang YC, Zhu ZW, Masuzaki S, Hasegawa H et al. Suppression of cell-cycle progression by Jun dimerization protein-2 (JDP2) involves downregulation of cyclin-A2. Oncogene 2010; 29: 6245–6256.
Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010; 467: 338–342.
Uren AG, Kool J, Berns A, van Lohuizen M . Retroviral insertional mutagenesis: past, present and future. Oncogene 2005; 24: 7656–7672.
Piu F, Aronheim A, Katz S, Karin M . AP-1 repressor protein JDP-2: inhibition of UV-mediated apoptosis through p53 down-regulation. Mol Cell Biol 2001; 21: 3012–3024.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Acknowledgements
DJA was supported by Cancer Research UK and the Wellcome Trust. LvdW was supported by the Kay Kendall Leukaemia Fund. CDRE was supported by Consejo Nacional de Ciencia y Tecnología (CONACYT) and the Wellcome Trust. MJA and REM were supported by Cancer Research UK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
van der Weyden, L., Rust, A., McIntyre, R. et al. Jdp2 downregulates Trp53 transcription to promote leukaemogenesis in the context of Trp53 heterozygosity. Oncogene 32, 397–402 (2013). https://doi.org/10.1038/onc.2012.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2012.56
Keywords
This article is cited by
-
Prevention of tumor risk associated with the reprogramming of human pluripotent stem cells
Journal of Experimental & Clinical Cancer Research (2020)
-
miR-501 acts as an independent prognostic factor that promotes the epithelial–mesenchymal transition through targeting JDP2 in hepatocellular carcinoma
Human Cell (2019)
-
Using PU.1 and Jun dimerization protein 2 transcription factor expression in myelodysplastic syndromes to predict treatment response and leukaemia transformation
Annals of Hematology (2019)
-
Sequencing methods and datasets to improve functional interpretation of sleeping beauty mutagenesis screens
BMC Genomics (2014)
-
Jun dimerization protein 2 is a critical component of the Nrf2/MafK complex regulating the response to ROS homeostasis
Cell Death & Disease (2013)
