
Abstract
Baculovirus is a promising vector for transducing numerous types of mammalian cells. We have developed hybrid baculovirus vectors and protocols for the efficient transduction of a variety of cell lines, primary cells and stem cells, including bone marrow–derived mesenchymal stem cells (BMSCs) and adipose-derived stem cells (ASCs). The hybrid vector enables intracellular minicircle formation and prolongs transgene expression. The advantages of this transduction protocol are that baculovirus supernatant alone needs to be added to cells growing in medium, and transduction occurs after only 4–6 h of incubation at room temperature (25 °C) with gentle shaking. The entire procedure, from virus generation to transduction, can be completed within 4 weeks. Compared with other transduction procedures, this protocol is simple and can confer efficiencies >95% for many cell types. This protocol has potential applications in tissue regeneration, as transduced cells continue to express transgenes after implantation. For example, transduction of rabbit ASCs (rASCs) with growth factor–encoding hybrid baculovirus vectors, as described as an example application in this protocol, enables robust and sustained growth factor expression, stimulates stem cell differentiation and augments tissue regeneration after implantation.
Similar content being viewed by others


Introduction
Baculovirus (Autographa californica multiple nucleopolyhedrovirus, AcMNPV) is an insect enveloped virus and is highly infectious to cultured insect cells, and thus baculovirus is often genetically engineered to infect host insect cells for recombinant protein production. Since it was found that baculovirus is able to transduce mammalian cells1,2, numerous permissive cells of human, rodent, porcine, canine, avian, feline and rabbit origins have been identified (for a review, see refs. 3,4,5,6). Baculovirus can transduce human embryonic stem cells7, mouse induced pluripotent stem (iPS) cells8 and mammalian cells cultured in 3D matrix9 and in suspension10. Within the baculovirus-transduced cells, transgenes can be expressed as long as they are driven by an appropriate promoter (e.g., cytomegalovirus (CMV) immediate-early or hybrid CAG promoter). Therefore, baculovirus has captured growing attention as a gene delivery vector for a plethora of applications, ranging from cell-based assay development11, protein production12, virus production10, virus-like particle production9, eukaryotic protein display13, RNA interference14, vaccine development15,16 and cancer therapy17 to tissue engineering of bone18,19, cartilage20,21 and heart22.
Comparison of baculovirus with other viral gene delivery systems
In comparison with other viral gene delivery vectors such as retrovirus, lentivirus, adenovirus and adeno-associated virus (AAV), baculovirus has a number of desirable features:
-
The natural host of baculovirus is insect cells, and thus baculovirus is nonpathogenic to humans and baculovirus neither replicates nor is toxic inside transduced mammalian cells23.
-
Baculoviral DNA degrades in the cells over time24,25, and baculoviral DNA integration into host chromosomes is extremely rare and cannot be found26,27 unless selective pressure is applied28.
-
Humans do not possess pre-existing antibodies29 and T cells30 specifically against baculovirus, which enables baculovirus to circumvent the pre-existing immunity problem.
-
Recombinant baculovirus can be readily constructed and propagated to high titers by infecting its natural host insect cells in biosafety level 1 (BSL-1) facilities4.
-
The baculovirus genome is large (∼134 kb) and hence it confers a huge cloning capacity of at least 38 kb (ref. 31).
The first three features ease safety concerns regarding the use of baculovirus for gene delivery, whereas the last two features simplify baculovirus vector production and expand the potential applications of baculovirus vectors.
However, there are three possible drawbacks associated with baculovirus vectors:
-
Owing to the nonreplication and nonintegration nature, baculovirus typically mediates transient (<7 d (refs. 3,4,5,6)) expression, which may hamper the applications of baculovirus vectors in some scenarios requiring long-term, stable expression (e.g., hemophilia and X-linked severe combined immunodeficiency).
-
Baculovirus is an enveloped virus and the baculoviral enveloped protein gp64 is essential for infection of insect cells and transduction of mammalian cells32. Consequently, baculovirus is vulnerable to mechanical forces, and bioprocessing steps during the vector purification (e.g., ultracentrifugation and filtration) tend to attenuate the virus titer.
-
Baculovirus is inactivated by serum complement system33, which may hamper the baculovirus administration by direct injection.
The advantages and disadvantages of baculovirus compared with other viral gene delivery vectors are further summarized in Table 1.
Key differences between our baculovirus transduction protocol and other protocols
Incubation with unconcentrated virus. For baculovirus transduction of permissive cells, frequently baculovirus supernatant is concentrated by ultracentrifugation and resuspended in PBS. Cells are then incubated with the virus for 1 h at 37 °C, using growth medium (e.g., DMEM) as the surrounding solution to adjust the final liquid volume2,34,35,36,37. However, ultracentrifugation may result in considerable loss in virus titer. Moreover, larger transduction processes require larger amounts of baculovirus, yet virus preparation by ultracentrifugation constitutes a formidable task upon process scale-up.
To simplify the transduction process, in our protocol cells are incubated with unconcentrated virus (i.e., virus supernatant harvested from the infected insect cell culture) at a lower temperature (e.g., 25 °C or 27 °C) for 4–8 h with mild shaking, using PBS as the surrounding solution24,38. This protocol enables and improves baculovirus-mediated gene transfer into HeLa38,39, human BMSCs40, BMSC-derived adipogenic progenitor cells41 and primary chondrocytes derived from rats24 and rabbits21,26, with efficiencies comparable or superior to those using concentrated virus. For instance, the transduction efficiencies of human BMSCs derived from umbilical cord blood can be elevated from ∼42% (using concentrated virus) to ∼73% (using our protocol)40. Our protocol also ameliorates AcMNPV-mediated transduction of BHK9, Vero39, canine MDCK42 and chicken Df-1 cells42, as well as augments the transduction of HEK293 and Schwann cells mediated by another baculovirus, Bombyx mori NPV43. Because our protocol obviates the need for virus ultracentrifugation, it represents a simpler approach and also considerably reduces possible virus inactivation during ultracentrifugation.
Surrounding solution during transduction. Baculovirus titer often falls in the range of 3 × 108 to 1 × 109 plaque-forming units per milliliter (pfu/ml). Transduction of cells cultured in six-well plates (e.g., 3 × 105 cells per ml) at a multiplicity of infection (MOI) of 100 (pfu per cell) only requires 30–100 μl of virus supernatant, and thus the virus supernatant needs to be diluted. For transduction in six-well plates, at least 500 μl of solution is required during the incubation period. Thus, virus supernatant can be diluted with insect cell medium to 100 μl and then mixed with the surrounding solution (400 μl) to a final volume of 500 μl. The volumetric ratio of diluted virus and surrounding solution can vary from 1:3 to 1:10, but in general a ratio of 1:4 is preferred (L.-Y.S., unpublished data).
The choice of surrounding solution has a considerable effect on transduction efficiency. We found that PBS as the surrounding solution confers better transduction efficiency and transgene expression than TNM-FH (the medium for baculovirus production) or DMEM38,40. Comparison between the major components in PBS and these media reveals that NaHCO3 present in DMEM and TNM-FH markedly inhibits baculovirus transduction by reducing the intracellular virus number44.
NaHCO3 is a buffering agent that is commonly added to the medium during medium preparation. Owing to the key role of NaHCO3 and concerns that incubation of certain cells (e.g., stem cells) in PBS for hours may damage the cells, we modified the protocol by using NaHCO3-deficient medium in lieu of PBS as the surrounding solution. By using our protocol, baculovirus is able to transduce various cell lines (e.g., Huh-7, HeLa and BHK), BMSCs27, ASCs from humans (hASCs)45 and rabbits (rASCs)46 and even cell sheets derived from rASCs22, at efficiencies exceeding 95%.
Temperature and length of transduction. Temperature and the length of time that cells are incubated with baculovirus also dictate the transduction efficiency. Recombinant baculovirus is produced by infecting insect cells that are cultured using insect cell medium (e.g., TNM-FH) at 27 °C. We found that incubation of baculovirus and mammalian cells at 25 °C or 27 °C results in better transduction than at 4 °C or 37 °C, because the physical integrity of enveloped baculovirus is compromised at 37 °C, whereas virus particle adsorption to cell surface is less efficient at 4 °C (ref. 38). Incubation of cells with a virus supernatant for a prolonged period of time can enhance the transduction efficiency and transgene expression, as longer incubation time enables better virus entry38. However, excessive incubation time at 27 °C may undermine cell health, and the optimal incubation time varies for different cells. In general, incubation for 4–6 h is recommended for most cell types24.
Other parameters influencing baculovirus transduction efficiency
Reagents enhancing transgene expression. Baculovirus-mediated transgene expression can be elevated by the addition of histone deacetylase (HDAC) inhibitors such as sodium butyrate37. HDAC inhibitors relax the chromatin structure by inducing histone hyperacetylation, which underscores the importance of epigenetic status of baculovirus genome for transgene expression. Notably, these drugs exert cytotoxicity at high concentrations47, and the extent to which gene expression is upregulated hinges on cell lines48. Therefore, optimal sodium butyrate concentration needs to be pre-determined for different cells.
Choice of promoter. Transgene expression is also promoter-dependent. Viral promoters that are active in mammalian cells, such as SV40, CMV, RSV and the hybrid CAG promoter, have been used to drive baculovirus-mediated gene expression with different strengths (for a review, see ref. 4). In addition, baculovirus-mediated transgene expression can be augmented by appending woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) to the 3′ end of transgene49. WPRE is a cis-acting RNA element that can stabilize mRNA and facilitate mRNA nuclear processing and export of mRNA into the cytoplasm, and thus it enhances transgene expression. A three- to tenfold increase in baculovirus-mediated enhanced green fluorescent protein (EGFP) expression is noted in several cell lines49.
Hybrid baculovirus vectors for prolonged transgene expression.
As mentioned above, baculovirus mediates transient transgene expression (typically <7 d), which may preclude the applications of baculovirus in certain scenarios requiring long-term expression. To prolong the expression, attempts to incorporate AAV inverted terminal repeats7,34 or the Sleeping Beauty transposon14,50 into baculovirus vectors have been made. These elements result in transgene integration into the host chromosome, and they can be used for sustained expression in neuronal cells and cancer cells.
In addition to developing integrating vectors, we have developed baculovirus systems that enable episomal maintenance of transgenes. Development of these systems is based on the hypothesis that shedding transgene cargo from the baculovirus genome to form an extrachromosomal DNA minicircle may prolong the transgene expression. With this assumption in mind, we established an FLP/Frt-based hybrid baculovirus system that comprises one baculovirus-expressing FLP recombinase and one substrate baculovirus harboring the transgene cassette flanked by two Frt sequences27. Co-transduction with the two baculovirus vectors successfully extended the transgene expression in a number of mammalian cells, including rBMSCs27 and rASCs51. The expression level and duration positively correlate with the recombination efficiency, presumably because minicircles confer stronger and longer transgene expression than their plasmid counterparts52. However, the FLP/Frt-mediated DNA minicircle formation occurs in only ∼40–50% of rBMSCs and rASCs51,53. To further enhance the recombination efficiency, we have developed a new FLPo/Frt system45 that explores the codon-optimized FLP (FLPo). The FLPo/Frt system enables recombination and minicircle formation in different mammalian cells, rASCs and rBMSCs at efficiencies >90%, and it has been successfully used in rASCs to extend and promote the expression of growth factors such as bone morphogenetic protein 2 (BMP-2)54, BMP-6 (ref. 20), transforming growth factor β3 (ref. 20) and vascular endothelial growth factor (VEGF)22.
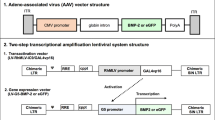
The new FLPo/Frt-based baculovirus system similarly consists of two baculoviruses: Bac-FLPo-expressing FLPo and the substrate baculovirus harboring the Frt-flanking transgene cassette (Fig. 1a). After co-transduction of cells, the expressed FLPo recognizes the Frt sites and excises the Frt-flanking cassette off the baculovirus genome, and hence it catalyzes the recombination and formation of episomal DNA minicircles encompassing the transgene cassette (Fig. 1b). The minicircle can persist in the cells for a longer term, whereas the baculovirus genome is rapidly degraded45. Aside from FLPo, two other site-specific recombinases, Cre and codon-optimized ΦC31 (ΦC31o), have been explored45. Cre catalyzes excision/recombination events between two identical loxP sites55, whereas ΦC31o mediates excision/recombination between heterotypic sites attP and attB. Similarly to our work with the FLPo/Frt-based baculovirus system, we constructed a binary baculovirus vector system based on Cre/loxP (Fig. 1a) and ΦC31o/attP/attB (not shown). Upon co-transduction, the transgene cassette in the substrate baculovirus is excised by the recombinase (ΦC31o or Cre) expressed by a second baculovirus vector, and it recombines into minicircles45.

(a) Illustration of the FLPo/Frt- and Cre/loxP sytems. (b) Illustration of the FLPo-mediated recombination and DNA minicircle formation. After co-transduction with Bac-FLPo and the substrate baculovirus, the expressed FLPo recognizes the Frt sites flanking the transgene cassette, leading to the excision of the transgene cassette, recombination and the formation of the DNA minicircle. CMV, cytomegalovirus immediate early promoter; Wp, WPRE element; pA, polyadenylation signal.
Experimental design
Gene delivery into mammalian cells using baculovirus requires multiple stages: (i) construction of a transgene-containing donor plasmid, (ii) generation of recombinant bacmid, (iii) generation of recombinant baculovirus, (iv) virus amplification and (v) transduction. The construction of the donor plasmid is discussed here, but it is omitted from the procedure.
Construction of the donor plasmid. Baculovirus naturally infects insect cells, and recombinant baculoviruses are traditionally constructed by homologous recombination in two steps. A transgene is first cloned into a transfer plasmid under the control of a native baculovirus promoter (e.g., polh or p10). The second step involves co-transfection of the plasmid and linearized parental baculovirus DNA into insect cells, in which the overlapping sequences on the transfer plasmid and parental DNA lead to homologous recombination and hence transgene insertion into the baculoviral genome. Subsequent viral replication generates the recombinant baculovirus containing an integrated copy of the transgene. This baculovirus construction system is commercially available (e.g., BacPAK, Clontech).
To generate baculovirus vectors for transduction, we alternatively use the Bac-To-Bac system (Invitrogen), which exploits site-specific transposition of transgene cassette on the donor plasmid into the baculovirus shuttle vector (bacmid) in E. coli. The system offers a series of donor plasmids such as pFastBac 1 and pFastBac DUAL. The plasmids also contain the baculovirus promoter (e.g., polh or p10) upstream of the multiple cloning site and flanking attachment sites Tn7R and Tn7L. For expression in mammalian cells, the baculovirus promoter is simply replaced by a promoter (e.g., CMV) that is active in mammalian cells using standard cloning techniques56 (designated as pBac-CMV5; Fig. 2), followed by cloning of the transgene into the donor plasmid.

The baculovirus promoter in the pFastBac vector is replaced by the CMV promoter to yield pBac-CMV5, and the transgene is cloned into the plasmid. The resultant donor plasmid is transformed into E. coli DH10Bac, in which the transposase expressed by the helper plasmid transposes the Tn7-flanking cassette from the donor plasmid to the Tn7 attachment site on the bacmid, resulting in transgene integration into the bacmid and disruption of the lacZα gene. In the agar plate containing antibiotics, IPTG and X-gal, the recombinant bacmid can be isolated from the white colonies. The recombinant bacmid is transfected into insect cells, in which the recombinant bacmid results in recombinant virus generation.
To generate the FLPo/Frt-based hybrid baculovirus system, construction of two baculoviruses is required: Bac-FLPo and the substrate baculovirus harboring the Frt-flanking transgene cassette (Fig. 1a). Construction of the FLPo-expressing vector (Bac-FLPo) is straightforward, simply by cloning the optimized flpo gene from pPGKFLPobpA into the donor plasmid (pBac-CMV5) under the control of the CMV promoter. To construct the substrate vector harboring a transgene, a DNA fragment composed of a multiple cloning site flanked by two Frt sites is PCR-amplified from pLOI2226 (ref. 57). The amplicon (0.25 kb) is subcloned into pFastBac DUAL to yield pBac-Frt. Second, a CMV promoter (0.6 kb) is PCR-amplified from pcDNA3.1(+) and subcloned into pBac-Frt to yield pBac-Frt-CMV. Third, a WPRE sequence and an SV40pA are cloned into pBac-Frt-CMV to generate pBac-FCW. Fourth, the transgene is subcloned into pBac-FCW downstream of the CMV promoter.
To assay the recombination efficiency mediated by either ΦC31o, Cre or FLPo, we also generated a substrate baculovirus Bac-ALF, which harbors d2egfp (encoding destabilized EGFP, d2EGFP) flanked by tandem recombination sites (attP-loxP-Frt and attB-loxP-Frt)45. To construct the donor plasmid pBacALF, the DNA fragment encoding the tandem recombination sites (XhoI-attP-loxP-Frt-BamHI-StuI-attB-loxP-Frt-HindIII) is first chemically synthesized and subcloned into pFastBac Dual to yield pALF. Second, the d2egfp-SV40pA fragment is PCR-amplified from pd2EGFP-N1 and inserted into the BamHI site of pALF to form pALF-dE. Finally, the CMV promoter (flanked by StuI/SmaI) is PCR-amplified from pcDNA3.1(+) and inserted into the StuI site of pALF-dE to yield pBacALF.
Generation of recombinant bacmid (Steps 1–33). To generate a recombinant bacmid, a donor plasmid is transformed into E. coli DH10Bac, which contains the bacmid and helper plasmid. The bacmid possesses a low-copy-number mini-F replicon (ori), a kanamycin resistance gene and a Tn7 attachment (att) site in frame within the lacZα gene. The helper plasmid within DH10Bac confers resistance to tetracycline and expresses the transposase (Fig. 2). After transformation of the recombinant donor plasmid into DH10Bac, the transposase transposes the Tn7-flanking cassette from the donor plasmid to the Tn7 attachment site, resulting in transgene integration into the bacmid and disruption of lacZα gene. The antibiotic selection and disruption of lacZα gene after transposition enable rapid selection/isolation of the recombinant bacmid from DH10Bac cultures. The recombinant bacmid is PCR-confirmed for the presence of transgene.
Production of recombinant baculovirus (Steps 33–44). The recombinant bacmid is used to transfect insect cells (e.g., Sf-9 derived from Spodoptera frugiperda), which leads to virus replication. Viral stocks collected from the transfected cells are defined as passage 0 (P0).
Amplification of recombinant baculovirus (Steps 45–56). The P0 baculovirus stocks can be used to further infect fresh Sf-9 cells for P1 virus production (Fig. 2). P1 virus can be used as an inoculum to infect Sf-9 cells for the generation of P2 virus stock. The resultant baculovirus is titrated by end-point dilution method58 (Box 1). To evaluate transduction efficiency, baculovirus vectors expressing EGFP47 and dsRed8 have been generated.
Transduction of mammalian cells (Step 57). In the PROCEDURE, we describe how to transduce cell lines (Step 57A, describing transduction of HEK293 as an example), iPS cells (Step 57B), rASCs (Step 57C) and a rASC sheet (Step 57D). The procedures for HEK293 and iPS cell culture are routine, and they have been described previously (see refs. 59 and 60 for iPS cells); thus, this protocol starts from the cell seeding. rASCs can be harvested s.c. from the inguinal fat pads surrounding the epididymis of 3–4-month-old New Zealand white (NZW) rabbits (see Box 2 for a detailed protocol)51.
For transduction, cells are seeded on to culture plates or flasks (e.g., six-well plates, 10-cm dishes, T-75 or T-150 flasks) and incubated overnight. At day 2, a certain volume (depending on the multiplicity of infection (MOI) and baculovirus titer) of unconcentrated baculovirus supernatant is diluted with fresh insect cell (Sf-9) culture medium, and it is then mixed with NaHCO3-free culture medium (e.g., DMEM or α-MEM) at a volumetric ratio of 1:4. The final volume of virus solution should be sufficient to wet the surface. Larger volumes of virus solution may reduce the chance of virus contact with cells and may decrease the transduction efficiency. Transduction is initiated by directly adding the appropriate volume (e.g., 500 μl per well in six-well plates, 4 ml per 10-cm dish and 5 ml per T-75 flask) of virus solution to cells. The transduction continues by gentle shaking on a rocking plate at room temperature for 4–6 h. After the transduction period, virus solution is replaced by the culture medium containing sodium butyrate. After ∼15–24 h of incubation at 37 °C, the medium is replaced by fresh medium. At day 3 (1 d post transduction, 1 dpt), cells are collected for analysis or applications (Fig. 3). For instance, cells transduced with Bac-CE (expressing EGFP) can be harvested at 1 dpt and analyzed by flow cytometry for the percentage of GFP+ cells (i.e., transduction efficiency). Alternatively, the cells continue to be cultured with medium exchange every 2–3 d (Fig. 3). For mock-transduction control, the procedure is the same, except that insect cell culture medium is used in lieu of virus supernatant.

Transduction of cells in a six-well plate.


Limitations of the protocol
The Bac-to-Bac system we commonly use eliminates the need for multiple rounds of plaque purification, which reduces the time required to obtain the P0 recombinant virus. However, the transgene in the genome of bacmid-derived baculovirus is more prone to spontaneous excision, leading to a rapid decrease in transgene expression in passages beyond P8 (ref. 61). Therefore, it is advised to keep the virus passage low.
Baculovirus transduction is highly efficient for a wide variety of cell types, yet it is reported that baculovirus transduction of hematopoietic cell lines such as U937, K562, Raw264.7, LCL-cm and Raji is inefficient (for a review, see ref. 6). In addition, baculovirus transduction efficiency for primitive stem cells such as embryonic stem cells62 and iPS cells8 is relatively low (<40–50%). Furthermore, baculovirus-mediated growth factor expression is relatively inefficient in rat or mouse ASCs or BMSCs (K.-C.L. and C.-L.Y., unpublished data), although the expression is robust in rASCs or rBMSCs53,54.
Last, although the transgene expression can be prolonged from <7 d to ∼3 weeks using the FLPo/Frt-based hybrid baculovirus vectors, for longer transgene expression (months to years) integration of transgene cassette is required. In this regard, hybrid baculovirus vectors by incorporating AAV inverted-terminal repeats7,34 or Sleeping Beauty transposons14,50 have been developed to enable transgene integration into the chromosome and long-term expression.
Materials
REAGENTS
-
MAX Efficiency DH10Bac chemically competent cells (Invitrogen, cat. no. 10361-012)
-
Sf-9 cells (ATCC, cat. no. CRL-1711)
-
Mammalian cells to be transduced, for example, HEK293 cells (ATCC, cat. no. CRL-1573), iPS cells (we have used the mouse iPS cell line 20D17, supplied by S. Yamanaka, Center for iPS Cell Research and Application, Kyoto University) or rASCs (derived as described in Box 2). See INTRODUCTION for a full discussion of the cell types that we have successfully transduced
-
S.O.C. medium (Invitrogen, cat. no. 15544-034)
-
LB broth (BD Biosciences, cat. no. 244620)
-
Agar (bacteriological; OXOID, cat. no. LP0011)
-
Sf-900 II serum-free medium (SFM; Invitrogen, cat. no. 10902-088)
-
TNM-FH insect cell medium (Sigma, cat. no. T3285)
-
DMEM (high glucose; Sigma, cat. no. D5648)
-
DMEM (low glucose; Gibco, cat. no. 31600-034)
-
α-MEM (Gibco, cat. no. 12000-022)
-
PBS without calcium or magnesium (pH 7.4; Invitrogen, cat. no. 14190-094)
-
Hyclone FBS (Thermo Scientific, cat. no. SH30070.03)
-
Sodium bicarbonate (NaHCO3; Sigma, cat. no. S5761)
-
Gentamycin (AMRESCO, cat. no. 0304)
-
Kanamycin (AMRESCO, cat. no. 0408)
-
Tetracycline (AMRESCO, cat. no. 0422)
-
X-gal (AMRESCO, cat. no. 0428)
-
IPTG (AMRESCO, cat. no. 0487)
-
Absolute 2-propanol (Sigma, cat. no. I9516)
-
Cellfectin II reagent (Invitrogen, cat. no. 10362-100)
-
Trypsin-EDTA (0.05% (wt/vol); Gibco, cat. no. 25300)
-
Tris-HCl (Sigma, cat. no. T5941)
-
EDTA (Sigma, cat. no. E6758)
-
Sodium hydroxide (NaOH; Sigma, cat. no. S8045)
-
SDS (Sigma, cat. no. L3771)
-
Potassium acetate (Sigma, cat. no. P1190)
-
DMSO (Sigma, cat. no. D2650)
-
Trypan blue stain (0.4%; APOLO, cat. no. APL-0167)
-
Ethanol (70% (vol/vol); Sigma, cat. no. 02877)
-
Gelatin (0.1% (wt/vol) solution) from porcine skin type A (Sigma, cat. no. G1890)
-
MEM non-essential amino acids solution, 100× (Gibco, cat. no. 11140-050)
-
2-mercaptoethanol (55 mM; Gibco, cat. no. 21985-023)
-
Leukemia inhibitory factor (107 U/ml; LIF, Millipore, cat. no. ESG1107)
-
Type I collagenase (Gibco, cat. no. 17100-017)
-
RNase A (Sigma, cat. no. R6513)
-
Penicillin-streptomycin, 100× (10,000 U/ml; Gibco, cat. no. 15140-122)
-
Penicillin-streptomycin-amphotericin B solution (Biological Industries, cat. no. 03-033-1B)
-
Sodium butyrate (Sigma, cat. no. B5887)
-
pFastBac vectors such as pFastBac 1 (Invitrogen, cat. no. 10360-014) and pFastBac Dual (Invitrogen, cat. no. 10712-024)
-
Recombinant baculovirus expressing EGFP (Bac-CE), DsRed (Bac-ER), FLPo (Bac-FLPo) and BMP-2 (Bac-FCBW) under the control of the CMV promoter
EQUIPMENT
-
Incubator, set at 37 °C, 5% CO2 (Thermo Scientific)
-
Incubator, 27 °C, no CO2 (Firstek Scientific, RI-102)
-
Shaking incubator, 27 °C, no CO2 (Cocono)
-
Upright ultra-low-temperature freezers, set at −80 °C (Forma 900 series, Thermo Scientific)
-
Refrigerator, 4 °C (Daytime)
-
Freezer, −20 °C (Elcold)
-
Water bath (Firstek Scientific)
-
Rotator (TAITEC, RT-50)
-
Laminar flow cabinet (UBI)
-
Sterilizer (Tomin, High-Pressure Steam Sterilizer)
-
Inverted microscope equipped with a phase-contrast ring and fluorescence filters (Nikon, TE200)
-
Upright microscope (Nikon)
-
Microtube (1.5 ml; Axygen, MCT-150-C)
-
Tubes (15 and 50 ml; Corning, cat. nos. 43079 and 35276, respectively)
-
Disposable culture tube (Kimble Chase, cat. no. 73500-16100)
-
Petri dish (Protech, cat. no. PT-PD-90)
-
Tissue culture plates (6-, 12- and 96-well; Thermo Scientific, cat. nos. 140675, 167008 and 150628, respectively)
-
Tissue culture flasks (T-75 and T-150; Thermo Scientific, cat. nos. 156499 and 159910, respectively)
-
Tissue culture centrifuge (Hettich Universal 32)
-
Microcentrifuge (Hettich Zentrifugen MIKRO-120)
-
Flow cytometer (BD Biosciences, FACSCalibur)
-
Rocking plate (TKS, RS01)
-
Vortex mixer (Scientific Industries, cat. no. SI-0236)
-
Multi-place magnetic stirrer (Thermolyne, Cellgro 45700)
-
Spinner flask (250 ml; Bellco, cat. no. 1965-80255)
-
Hemocytometer (Marienfeld-Superior, cat. no. 6300-30)
-
Pipettes (5 and 10 ml; Costar, cat. nos. 4487 and 4488, respectively)
-
Tips (10, 200 and 1,000 μl; MultiMax, cat. nos. 7230, 2947 and 2950, respectively)
-
Pipettors (P2N, P10N, P20N, P200N and P1000N; Gilson, cat. nos. F144561, F144562, F144563, F144565 and F144566, respectively)
-
Pipette aid
-
Surgical scissors
-
Surgical forceps
-
Shaver
-
Scalpel
-
Induction chamber
-
Glass cell spreaders
-
Laboratory glass bottle (250 ml; Schott Duran, cat. no. 2180136)
-
Cryogenic vials (Nalgene, cat. no. 5000-0020)
-
Cryo box
-
Syringes (BD Biosciences, 1-ml syringe with a 26-G needle for anesthesia)
-
Bottle-top filters (0.22-μm; Sartorius, cat. no. 9.049 205)
-
Syringe filter (0.22-μm; Nalgene, cat. no. 190-2520)
REAGENT SETUP
LB broth
-
To prepare 1,000 ml of LB broth, dissolve 25 g of the LB broth powder in 1,000 ml of distilled water, and mix it thoroughly. Autoclave the medium at 121 °C for 15 min. Cool the broth to room temperature, and then store it at 4 °C for up to 3 months.
LB agar plate
-
Dissolve 25 g of LB broth powder and 15 g of agar powder in 1,000 ml of distilled water and mix it thoroughly. Autoclave the solution at 121 °C for 15 min. When the temperature of the heated solution drops to 55 °C, add gentamycin, kanamycin, tetracycline, X-gal and IPTG to final concentrations of 7, 50, 10, 200 and 40 μg/ml, respectively. Divide the agar mixture into separate Petri dishes (20 ml per dish) and cool the plates to room temperature. Store the plates at 4 °C for up to 1 month.
Resuspension buffer
-
To prepare 100 ml of resuspension buffer, dissolve 0.24 g of Tris-HCl and 0.29 g of EDTA in 80 ml of distilled water, and then adjust the pH to 8.0. Add 10 mg of RNase A and fill the container with distilled water to a final volume of 100 ml. Store the buffer at 4 °C and use it within 1 month.
Lysis buffer
-
To prepare 100 ml of lysis buffer, dissolve 0.8 g of NaOH and 1 g of SDS in 100 ml of distilled water. Store the buffer at room temperature and use it within 3 months. Mix the buffer thoroughly before use.
Critical
If precipitates form in the lysis buffer, warm the buffer in a 37 °C water bath until the precipitates totally dissolve.
Precipitation buffer
-
To prepare 100 ml of precipitation buffer, dissolve 29.4 g of potassium acetate in 50 ml of distilled water and adjust the pH to 5.5. Fill with distilled water to a final volume of 100 ml. Store the buffer at room temperature and use it within 3 months.
Elution buffer
-
To prepare 100 ml of elution buffer, dissolve 0.16 g of Tris-HCl and 0.03 g of EDTA in 80 ml of distilled water, adjust the pH to 8.0, and use distilled water to bring the final volume to 100 ml. Store the buffer at room temperature and use it within 1 month.
Sf-9 medium
-
To prepare 1,000 ml of Sf-9 medium, mix 900 ml of TNM-FH medium (filtered through 0.22-μm) with 100 ml of FBS. Store the medium at 4 °C for up to 1 month.
HEK293 medium
-
To prepare 500 ml of HEK293 medium, mix 450 ml of DMEM (high glucose) medium (filtered through a 0.22-μm filter) and 50 ml of FBS. Store the medium at 4 °C for up to 1 month.
Gelatin-coated culture flask and plate
-
Add 0.1% (wt/vol) gelatin solution to a T-75 flask (5 ml) or a six-well plate (1 ml). Ensure that the entire bottom surface is covered with liquid. Incubate the flask or plate at 37 °C for 30 min. Aspirate the gelatin solution before use. Prepare the flask or plate immediately before use.
iPS medium
-
To prepare 500 ml of iPS medium, mix 419 ml of DMEM (high glucose) medium (filtered through a 0.22-μm filter), 75 ml of FBS (15% (vol/vol), 5 ml of 100× non-essential amino acids, 0.9 ml of 2-mercaptoethanol and 50 μl of LIF. Store the medium at 4 °C and use it within 1 month.
rASC medium
-
rASC medium can be DMEM- or α-MEM–based. To prepare 500 ml of rASC medium, mix 445 ml of DMEM (low glucose) medium or α-MEM medium (filtered through a 0.22-μm filter), 50 ml of FBS (10% (vol/vol) and 5 ml of 100× penicillin-streptomycin. Store the medium at 4 °C for up to 1 month.
rASC sheet fabrication medium
-
To prepare 500 ml of rASC sheet fabrication medium, mix 395 ml of α-MEM medium (filtered through a 0.22-μm filter), 100 ml of FBS (20% (vol/vol)) and 5 ml of 100× penicillin-streptomycin. Store the medium at 4 °C for up to 1 month.
Surrounding solution
-
Surrounding solution is culture medium containing 10% (vol/vol) FBS, without NaHCO3. The surrounding medium should be stored at 4 °C and used within 1 month.
Critical
The surrounding medium is prepared in a similar manner to the culture medium, except that 3.7 grams per liter NaHCO3 is not added.
Type I collagenase solution
-
To prepare 5 ml of 2% (wt/vol) type I collagenase, dissolve 0.1 g of type I collagenase powder in a final volume of 5 ml of PBS solution. Type I collagenase solution should be prepared freshly before starting the isolation process, and it should be filter-sterilized using a 0.22-μm syringe filter.
Sodium butyrate stock solution
-
To prepare 100× stock solution of sodium butyrate (300 mM), dissolve 1 g of sodium butyrate in 30.3 ml of distilled water and sterilize the solution by passing it through a 0.22-μm filter. Freeze the stock solution in aliquots at −20 °C and use it within 6 months.
Procedure
Generation of recombinant bacmid
Timing 5 d
Critical Step
If a recombinant baculovirus stock is already available, Steps 1–44 can be skipped.
-
1
Transposition of pFastBac donor plasmid, day 1. Dilute the donor plasmid to 10 ng/μl using sterile water in a sterile 1.5-ml microtube.
-
2
Thaw one vial of MAX Efficiency DH10Bac competent cells on ice.
-
3
Prechill a sterile 1.5-ml microtube on ice.
-
4
Add 30 μl of DH10Bac competent cells into the prechilled microtube and keep it on ice.
-
5
Add 1 μl of the diluted donor plasmid into 30 μl of DH10Bac competent cells.
-
6
Incubate the competent cells on ice for 20 min.
Critical Step
Do not hold the bottom of the microtube with your finger. Hold the upper part of the microtube to avoid warming the competent cells.
-
7
Incubate the mixture in the 42 °C water bath for 60 s to perform heat shock.
-
8
Immediately transfer the mixture to ice and leave it on ice for 5 min.
-
9
Add 500 μl of S.O.C. medium that has been prewarmed to room temperature into the microtube containing competent cells. Shake the tube in a rotator (250 r.p.m.) at 37 °C for 4 h. Thirty minutes before the end of this 4-h incubation, prewarm an LB agar plate at 37 °C for 30 min.
-
10
Centrifuge the cells at 9,000g for 2 min at room temperature. Remove 470 μl of supernatant (leaving 60 μl). Resuspend the cell pellet by pipetting gently.
-
11
Spread 60 μl of the resuspended competent cells on the prewarmed LB agar plate using a glass cell spreader.
-
12
Incubate the LB agar plate at 37 °C for 48 h for colony formation. Store the plate at 4 °C before the next step if necessary.
Critical Step
Normally, blue and white colonies are formed after incubation. Successful transgene cassette transposition into the attachment site disrupts the lacZα gene, leading to interrupted expression of LacZα peptide and the formation of white colonies. Without transposition, lacZα gene remains intact, giving rise to LacZα peptide expression and the formation of blue colonies in the presence of X-gal in the LB agar plate. Therefore, colonies containing the recombinant bacmid are white in a background of blue colonies that harbor the unaltered bacmid.
Critical Step
The process may be accelerated by incubating the agar plate for only 16–24 h. However, at this time, colonies are relatively small. For the following bacmid isolation, pick the largest, most isolated white colonies to avoid selecting false positives. Avoid picking colonies that appear gray or are darker in the center, as they can contain a mixture of cells with empty bacmid and recombinant bacmid.
Pause point
The LB agar plate can be stored at 4 °C for 1 month.
-
13
Day 3. Prewarm the spread LB agar plate to room temperature.
-
14
Prewarm another fresh LB agar plate at 37 °C for 30 min and divide the fresh LB agar plate into three areas with a marker.
-
15
Pick a blue colony (as control) and two white colonies onto the spread LB agar plate and re-streak them on the three areas of the fresh LB agar plate.
-
16
Incubate the re-streaked LB agar plate at 37 °C for 16–24 h for colony formation. Store the plate at 4 °C before the next step if necessary.
Critical Step
The white colonies should contain the recombinant bacmid. Verify the integration of the transgene cassette into bacmid by restriction analysis or PCR if necessary.
Pause point
The re-streaked LB agar plate can be stored at 4 °C for 1 month.
-
17
Isolation of recombinant bacmid DNA, day 4. Add 3 ml of LB broth containing gentamycin (7 μg/ml), kanamycin (50 μg/ml) and tetracycline (10 μg/ml) into a disposable culture tube.
-
18
Pick a white colony from the re-streaked LB agar plate, and then inoculate the cells into the disposable culture tube.
Critical Step
We recommend that you culture two white colonies individually in LB broth to ensure successful isolation of recombinant bacmids.
-
19
Place the culture tube on the rotator (250 r.p.m.) at 37 °C for 20 h.
-
20
Day 5. Transfer 1 ml of cultured bacterial cells to a 1.5-ml microtube and centrifuge the tube at 9,000g for 2 min at room temperature. Remove all of the supernatant.
-
21
Add 200 μl of resuspension buffer to the pellet. Resuspend the cells by pipetting or vortexing.
-
22
Add 200 μl of lysis buffer to the cell suspension. Mix it gently by inverting the microtube ten times.
-
23
Incubate the mixture at room temperature for 5 min.
-
24
Add 300 μl of precipitation buffer to the mixture. Mix it immediately by inverting the microtube ten times. Do not vortex.
-
25
Chill the microtube on ice for 10 min. Centrifuge the mixture at 15,000g at 4 °C for 20 min.
-
26
Transfer 600 μl of the supernatant to another 1.5-ml microtube.
Critical Step
Avoid pipetting any white precipitate in the supernatant. Pipette the clear supernatant into the microtube and centrifuge it at 15,000g for another 5 min at 4 °C to remove any contaminating precipitates.
-
27
Add 800 μl of absolute 2-propanol to the clear supernatant. Mix it gently by inverting the microtube ten times.
-
28
Chill the microtube on ice for 10 min. Centrifuge the tube at 15,000g at 4 °C for 15 min. Remove the supernatant.
-
29
Add 500 μl of 70% (vol/vol) ethanol to the pellet. Mix gently by inverting the microtube ten times.
-
30
Centrifuge the tube at 15,000g at 4 °C for 5 min. Remove the supernatant completely.
-
31
Open the cap of the microtube and air-dry the DNA pellet for 5–10 min until the pellet becomes half-transparent.
-
32
Resuspend the DNA pellet in 40 μl of elution buffer by gentle pipetting. Allow the pellet to dissolve completely on ice or in a 4 °C refrigerator.
Critical Step
To avoid shearing the bacmid DNA, do not pipette more than two times.
-
33
Divide the bacmid DNA into aliquots in separate sterile 1.5-ml microtubes, and store them at −20 °C to avoid more than one freeze-thaw cycle.
Pause point
The bacmid DNA can be stored at −20 °C for up to 2 weeks.
Production of recombinant baculovirus
Timing 5–7 d
-
34
Transfection of recombinant bacmid DNA, day 1. Grow Sf-9 cells with Sf-9 medium (containing 10% (vol/vol) FBS) in suspension to log-phase (e.g., 1–2 × 106 cells per ml) with >95% viability.
Critical Step
Sf-9 cells can be readily cultured in monolayer or in suspension at 27 °C without CO2.
-
35
Dilute Sf-9 cells to 3 × 105 cells per ml and seed 1 ml of cells to each well of a 12-well plate. Incubate the cells in a 27 °C incubator for at least 30 min to allow cell attachment.
-
36
For each bacmid that is ready for transfection, prepare solutions A and B (containing bacmid) in a sterile 1.5-ml microtube as follows:
Table 3
Solution A
Solution B
Reagent
Volume (μl)
Reagent
Volume (μl)
Cellfectin II reagent
2
Bacmid DNA
5
Sf-900 II SFM
30
Sf-900 II SFM
30
Critical Step
SFM is used in transfection because proteins in the FBS interfere with the Cellfectin reagent and inhibit the transfection.
-
37
Mix gently solutions A and B in a sterile 1.5-ml microtube, and incubate it at room temperature for 30 min.
-
38
Add 240 μl of Sf-900 II SFM to the DNA-lipid mixture and mix it thoroughly.
-
39
Wash the attached Sf-9 cells with Sf-900 II SFM twice to remove FBS. Discard the medium completely.
-
40
Add the DNA-lipid mixture to the 12-well plate (300 μl per well). Incubate the 12-well plate at 27 °C for 5 h for transfection.
Critical Step
Do not add antibiotics during transfection.
-
41
After transfection, discard the DNA-lipid mixture in the well. Add 1 ml of Sf-9 medium containing 10% (vol/vol) FBS and 1% (vol/vol) penicillin-streptomycin-amphotericin B to each well. Incubate the 12-well plate at 27 °C for 4–6 d.
-
42
Day 5–7. Visually observe the cytopathic effect (CPE) of transfected Sf-9 cells under a microscope. When CPE is observed in ∼30% of cells, collect the culture medium in a sterile 1.5-ml microtube.
Critical Step
At 3 d after transfection, visually check the CPE of transfected cells (e.g., cell lysis, for which cells appear lysed resulting in clearing plaques on the cell monolayer). If transfection efficiency is low, CPE may not be obvious until 4 or 5 d after transfection.
-
43
Centrifuge the culture medium at 500g for 10 min at room temperature. Pipette the supernatant into a sterile 1.5-ml cryogenic vial as the P0 virus.
-
44
Divide the P0 virus into separate sterile 1.5-ml cryogenic vials. Store the working stock at 4 °C and the master stock at −80 °C for long-term storage.
Critical Step
Do not store the virus stock at −20 °C, which leads to more rapid titer loss63. Protect the stock from light.
Pause point
The P0 virus can be stored for at least 3 months at 4°C and for years at −80 °C.
Amplification of recombinant baculovirus
Timing 10–12 d
-
45
Day 1. Grow Sf-9 cells using Sf-9 medium to log phase (∼1 × 106 cells per ml). Seed 10 ml of Sf-9 cells to a T-75 flask. Incubate the cells at 27 °C for at least 30 min to allow cell attachment.
Critical Step
For baculovirus amplification, Sf-9 medium containing 10% (vol/vol) FBS is recommended because baculovirus is more stable in the presence of FBS.
-
46
Aspirate 5 ml of medium from the T-75 flask to a 15-ml centrifuge tube. Add 25 μl of P0 virus stock to the medium and mix it thoroughly.
-
47
Transfer the 5-ml medium containing P0 virus back to the T-75 flask. Continue the infection by incubating the flask in a 27 °C humidified incubator for 4–5 d.
Critical Step
Infecting cells at low MOI (e.g., 0.05–0.1) is recommended for baculovirus amplification. Infecting cells with higher MOI may generate noninfectious defective virus particles and it may reduce the quality of the baculovirus stock. At this stage, we do not routinely determine the virus titer, because it takes 12–15 d. Instead, we assume a virus titer of 2 × 107 pfu/ml according to our experience, and add 25 μl of virus stock to the T-75 flask. Do not directly add the virus stock into the T-75 flask. We recommend premixing the virus stock with 5 ml of Sf-9 medium before infection.
-
48
Day 5 (−6). Check the CPE of infected Sf-9 cells. When CPE (e.g., cell lysis) is observed in ∼70% of cells, collect the culture supernatant in a sterile 15-ml centrifuge tube.
-
49
Centrifuge the cells at 500g for 10 min at room temperature. Pipette the supernatant into another sterile 15-ml centrifuge tube as the P1 virus stock.
-
50
Store the P1 virus stock at 4 °C and protect it from light. For long-term storage, transfer the virus stock into separate 1.5-ml cryogenic vials and store them at −80 °C.
Pause point
The P1 virus can be stored at −80 °C for years. When storing at 4 °C, the virus is stable for at least 3 months.
-
51
Day 6 (−7). To generate P2 virus stock, grow 200 ml of Sf-9 cells in the 250-ml spinner flask to log phase (∼1.0–1.2 × 106 cells per ml) using Sf-9 medium.
-
52
Transfer 10 ml of culture from the spinner flask to a 15-ml centrifuge tube. Add 50 μl of the P1 virus stock to the 10-ml culture.
Critical Step
The titer (pfu/ml) of baculovirus can be determined using either plaque assay58 or end-point dilution assay, which is described in detail in Box 1. If the virus titer is not determined at this stage, a virus inoculum volume of 50 μl (assuming a P1 virus titer of 2 × 108 pfu/ml) is recommended. Premix the virus stock with 10 ml of culture.
-
53
Transfer the 10-ml culture containing P1 virus back to the spinner flask. Place the spinner flask on the magnetic stirrer (100 r.p.m.) in a 27 °C humidified incubator and incubate it for 4–5 d.
-
54
Days 10–12. Check the viability of infected Sf-9 cells by trypan blue dye exclusion with a hemocytometer. When the cell viability drops to ∼70%, collect the culture in four sterile 50-ml centrifuge tubes.
-
55
Centrifuge at 500g for 10 min at room temperature. Transfer the supernatant to a sterile 250-ml laboratory glass bottle as the P2 virus stock.
-
56
Store the P2 virus stock at 4 °C and protect it from light. Titrate the virus as described in Box 1.
Critical Step
For mammalian cell transduction, in general, a batch of 100–200 ml of P2 virus stock is sufficient for experiments.
Pause point
The P2 virus can be stored at 4 °C for at least 3 months.
Transduction of mammalian cells with baculovirus
-
57
To transduce general cell lines (e.g., Huh-7, SNU-449, A549, HeLa and HEK293), follow option A. Option A describes the transduction of HEK293 cells in six-well plates using a baculovirus (Bac-CE) expressing EGFP as an example. To transduce iPS cells, follow option B, which describes the transduction of iPS cells using Bac-ER (containing a EF1α-DsRed transgene cassette) as an example. To transduce rASCs using the FLPo/Frt-based hybrid system, follow option C. To transduce rASC sheets, follow option D. Option D demonstrates the fabrication and transduction of rASC sheet using Bac-CE.
-
A
Transduction of a general cell line
-
i
Day 1. Trypsinize HEK293 cells from the T-75 flask and resuspend the cells with prewarmed HEK293 medium to a density of 1.5 × 105 cells per ml.
Critical Step
We prefer not to add antibiotics, as they mask bacterial contamination. Nonetheless, antibiotics do not affect baculovirus transduction, and they can be used in primary cells such as rASCs.
-
ii
Seed 2 ml of cell suspension into each well of a six-well plate (3 × 105 cells per well). Incubate the cells overnight at 37 °C. After overnight incubation, HEK293 cells should reach 60–70% confluence.
Critical Step
The initial seeding cell may slightly vary from cell line to cell line, depending on the cell size.
-
iii
Day 2. Calculate the required virus volume based on the virus titer (typically 3 × 108–1 × 109 pfu/ml for P2 virus) and cell number (3 × 105 cells per well). For each well, the virus volume required is calculated using the following formula: volume required (ml) = desired MOI (pfu per cell) × cell number (cells)/virus titer (pfu/ml).
-
iv
For each well, add the required volume of Bac-CE into Sf-9 medium to a total volume of 100 μl. Next, mix 100 μl of virus solution with 400 μl of surrounding solution (NaHCO3-free HEK293 medium).
Critical Step
For example, when 3 × 105 cells are transduced by Bac-CE with a titer of 5 × 108 pfu/ml at MOI 100, the volume required for each virus supernatant is 100 (pfu per cell) × 3 × 105 (cells)/5 × 108 (pfu/ml) = 0.06 ml (60 μl). Add 60 μl of Bac-CE to 40 μl of Sf-9 medium. Mix the 100 μl of virus mixture with 400 μl of surrounding solution. For mock transduction as control, simply mix 100 μl of Sf-9 medium with 400 μl of surrounding solution.
Critical Step
If the calculated total virus volume exceeds 100 μl, increase the surrounding solution volume with the fixed volumetric ratio (virus solution:surrounding solution = 1:4).
Critical Step
If a larger vessel (e.g., a T-75 or T-150 flask) is used, the volume of the final virus solution is proportionally increased according to the culture area.
-
v
Discard the HEK293 medium and wash the cells twice with PBS (1 ml per well).
-
vi
Remove PBS and initiate the transduction by adding 500 μl of the mixed virus solution into each well.
-
vii
Put the six-well plate on a rocking plate. Continue the transduction process by gently shaking the plate (10 times per min) for 6 h at room temperature.
Critical Step
Transduction efficiency depends on cell type, MOI and incubation time, which may vary from 4 to 6 h. For HEK293, transduction at MOI 100 for 6 h at room temperature yields transduction efficiency >95% without compromising cell viability. Incubation at 37 °C results in reduced transduction efficiency because baculovirus integrity is compromised at 37 °C.
-
viii
Aspirate the virus solution and wash the cells again. Add fresh HEK293 medium containing 3 mM sodium butyrate. Incubate the mixture at 37 °C for 24 h.
Critical Step
Sodium butyrate enhances transgene expression, but high sodium butyrate concentration is toxic to cells. We determine that 3 mM enhances the transduction of HEK293 without compromising cell viability.
-
ix
Day 3. Observe the fluorescence-emitting cells under the fluorescence microscope. Trypsinize and resuspend cells in 0.5 ml of PBS. Analyze the percentage of fluorescence-emitting cells using a flow cytometer.
Timing 3 d
-
i
-
B
Transduction of iPS cells
-
i
Day 1. Seed iPS cells to a gelatin-coated six-well plate (2 × 105 cells per well, 2 ml). Keep the cells at 37 °C overnight.
-
ii
Day 2. Calculate the required virus volume as described in Step 57A(iii).
-
iii
Remove the medium and gently wash the cells twice with PBS (1 ml per well).
-
iv
For each well, add the required volume of baculovirus (Bac-ER) into Sf-9 medium to a total volume of 100 μl. Next, mix the 100 μl of virus solution with 400 μl of surrounding solution (NaHCO3-free iPS medium).
Critical Step
The iPS cells express GFP under the control of the Nanog promoter. Therefore, here we transduce the cells with a baculovirus expressing DsRed (emitting red fluorescence).
-
v
Remove PBS from the six-well plate. Add the mixed virus solution to the wells (500 μl per well).
-
vi
Gently shake the six-well plate on a rocking plate (10 times per min). Continue the transduction process for 4 h at room temperature.
Critical Step
iPS cells do not adhere firmly to the plate. Treat the cells gently.
-
vii
After transduction, aspirate the baculovirus-containing solution and gently wash the cells twice with 1 ml of PBS. Replenish with 2 ml of iPS medium containing 3 mM sodium butyrate. Incubate the cells at 37 °C for 24 h.
-
viii
Day 3. Observe the DsRed expression under a fluorescence microscope. Trypsinize and resuspend the cells in 0.5 ml of PBS. Analyze the percentage of fluorescence-emitting cells using a flow cytometer (543 nm).
Timing 3 d
-
i
-
C
Transduction of rASCs
-
i
Day 1. Move the cryogenic vial containing rASCs (1 × 106 cells per vial) from liquid nitrogen. Thaw the cells in a 37 °C water bath. Be sure not to immerse the cap.
-
ii
Once the crystals melt (1–2 min), transfer the cells to a T-75 flask containing 9 ml of prewarmed rASC medium. Keep the cells at 37 °C in a 5% CO2 incubator.
-
iii
After 6 h of attachment, replace the DMSO-containing medium with 10 ml of rASC medium. Culture the cells at 37 °C for 2–3 d until ∼80–90% confluency.
Critical Step
As rASCs tend to differentiate upon 100% confluency, do not allow the cells to grow to full confluency. We recommend subculturing rASCs at ∼80–90% confluency.
-
iv
Day 3 (−4). Wash the cells with 10 ml of PBS twice, and discard the solution. Add 1 ml of prewarmed 0.05% (wt/vol) trypsin-EDTA to the T-75 flask and incubate it at 37 °C for 5 min to detach rASCs. Check for complete cell detachment by examining cell morphology under a microscope.
-
v
Inactivate trypsin by adding 5 ml of rASC medium and gently resuspending the cells. Aspirate 50 μl of cell solution for counting (the concentration is generally 4–6 × 105 cells per ml).
-
vi
Dilute the cell suspension to 1 × 105 cells per ml with rASC medium and seed 2 ml of cell solution to the six-well plate (2 × 105 cells per well or ∼2 × 104 cells per cm2). Incubate the cells at 37 °C overnight. After overnight incubation, the rASC density is ∼2 × 105 cells per well (∼60–70% confluency).
Critical Step
For rASC transduction in a larger scale, seed rASCs to T-75 or T-150 flasks with the same cell density (∼2 × 104 cells per cm2) and incubate them overnight in the same manner.
-
vii
Day 4 (−5). Calculate the required virus volume as described in Step 57A(iii). For example, when 2 × 105 cells are co-transduced by Bac-FLPo and the substrate baculovirus with the same titer (e.g., 5 × 108 pfu/ml) at the same MOI (e.g., 50), the volume required for each virus supernatant is 50 (pfu per cell) × 2 × 105 (cells)/5 × 108 (pfu/ml) = 0.02 ml (20 μl). Add 20 μl of Bac-FLPo and 20 μl of substrate baculovirus to 60 μl of Sf-9 medium. Mix the 100 μl of virus mixture with 400 μl of surrounding solution. For mock transduction as control, simply mix 100 μl of Sf-9 medium with 400 μl of surrounding solution.
-
viii
For each well, add the required volume of Bac-FLPo and substrate baculovirus into Sf-9 medium to a total volume of 100 μl. Next, mix the 100 μl of virus solution with 400 μl of surrounding solution (NaHCO3-free rASC medium).
-
ix
Discard rASC medium and wash the cells twice with PBS (1 ml per well). Remove the PBS.
-
x
Initiate the transduction by adding the mixed virus solution (500 μl) into each well.
-
xi
Put the six-well plate on a rocking plate. Continue the transduction process by gently shaking the plate (10 times/min) for 6 h at room temperature.
-
xii
After 6 h of transduction, remove the virus solution and add rASC medium (containing 3 mM sodium butyrate). Incubate the mixture at 37 °C for 24 h.
-
xiii
Day 5 (−6). At 1 dpt, replace the sodium butyrate-containing medium with fresh rASC medium.
Critical Step
Here we describe the co-transduction of rASCs cultured in a six-well plate with the FLPo/Frt-based hybrid baculovirus system (Bac-FLPo and its substrate baculovirus) as an example. This procedure starts from the thawing of the primary rASCs, isolated as described in Box 1.
Timing 5–6 d
-
i
-
D
Transduction of a rASC sheet
-
i
Day 1. Seed rASCs (prepared as described in Step 57C(i–iii)) to a six-well plate (5 × 105 cells per well) with 2 ml of rASC sheet fabrication medium. Keep the cells at 37 °C in a 5% CO2 incubator for 2 d.
Critical Step
To facilitate the cell sheet formation, higher seeding cell density (5 × 105 cells per well) is helpful. The 20% (vol/vol) of FBS in the fabrication medium also augments the secretion of extracellular matrix by rASCs to form the cell sheet.
-
ii
Day 3. The rASC sheet should be visible. Remove the medium and wash the cell sheet gently with PBS (2 ml per well) twice.
-
iii
Calculate the required virus volume.
-
iv
For each well, add the required volume of baculovirus (e.g., Bac-CE) into Sf-9 medium to a total volume of 100 μl. Next, mix the 100 μl of virus solution with 400 μl of surrounding solution (NaHCO3-free rASC medium).
-
v
Initiate the transduction process by adding the virus solution to wells (500 μl per well). Put the six-well plate on a rocking plate. Continue the transduction process by gently shaking the plate (10 times per min) for 6 h at room temperature.
-
vi
Terminate the transduction process by removing the virus solution.
-
vii
Add the rASC sheet fabrication medium containing 3 mM sodium butyrate (2 ml per well) and incubate it at 37 °C for 15 h.
-
viii
Day 4. Replace the medium with fresh rASC sheet fabrication medium and continue to incubate the cell sheet for 9 h.
-
ix
Wash the cell sheet with PBS twice and statically incubate with 0.05% (wt/vol) trypsin-EDTA (0.5 ml per well) for 10 s.
Critical Step
The time for trypsin-EDTA treatment is crucial. Longer treatment time (e.g., 2 min) may dissociate the cell sheet. For rASC sheet, 10 s is sufficient to detach the cell sheet without compromising the sheet integrity.
-
x
Remove trypsin-EDTA and wash the rASC sheet with PBS twice.
-
xi
Remove residual PBS and replenish with 2 ml of PBS. Gently shake the six-well plate by hand until the transparent rASC sheet spontaneously detaches from the well. In general, gentle shaking for <1 min is sufficient to detach the cell sheet.
Critical Step
To quantify the transduction efficiency, the cell sheet can be dissociated by prolonging the trypsin-EDTA treatment time to 5 min. The dissociated cells are analyzed by flow cytometry.
Timing 4 d
-
i
-
A
Troubleshooting
Troubleshooting advice can be found in Table 2.
Timing
Steps 1–33, generation of recombinant bacmid: 5 d
Steps 34–44, production of recombinant baculovirus (P0): 5–7 d
Steps 45–56, amplification of recombinant baculovirus (P2): 10–12 d
Step 57, transduction of mammalian cells with baculovirus: 3–6 d
Box 1, titration of recombinant baculovirus: 12–15 d
Box 2, isolation and culture of rASCs: 21–23 d
Anticipated results
The transduction procedures in Step 57 option A yield high transduction efficiencies and transgene expression levels in a wide variety of mammalian cell lines (Fig. 4). However, iPS cells are less permissive to baculovirus transduction. At MOI 100, the transduction efficiency was ∼45% using option B (data not shown). For recombination efficiency analysis, the cells were co-transduced with the recombinase-expressing baculovirus (Bac-FLPo or Bac-Cre) and the substrate baculovirus Bac-ALF and analyzed by flow cytometry at 1 dpt. Co-transduction resulted in recombination efficiencies approaching 90%–95% in various cell lines (e.g., Huh-7, HeLa, BHK) and stem cells such as rASCs (Fig. 5)45.

(a) Illustration of Bac-CE. (b) Fluorescence micrographs captured at 1 dpt. Magnification, 200×. Scale bars, 100 μm. (c) Transduction efficiency (TE) and total fluorescence intensity (FI). Huh-7, SNU-449, A549, HeLa and HEK293 cells were transduced with Bac-CE at MOI 100 and analyzed by flow cytometry. The data represent the means ± s.d. of three independent experiments.

(a) Illustration of Bac-ALF vector and recombined minicircle. Co-transduction of cells with the recombinase-expressing baculovirus and Bac-ALF results in the transgene excision and recombination to form an ∼2.2-kb minicircle and hence the placement of d2egfp downstream of the CMV promoter. Therefore, analysis of GFP+ cells yields recombination efficiency. (b) Comparison of Cre- and FLPo-mediated recombination in different mammalian cells. At 1 dpt, the d2EGFP expression was monitored with a fluorescence microscope. Scale bars, 100 μm. (c) Recombination efficiency analysis in different cells. HEK293, Huh-7, HeLa, BHK, RD, rASCs and hASCs were co-transduced with Bac-Cre or Bac-FLPo (MOI 50) and Bac-ALF (MOI 200). At 1 dpt, the cells were analyzed by flow cytometry. The data represent the means ± s.d. of three independent experiments. Panels b and c were adapted from ref. 45 under Creative Commons license 3.0.
rASCs were co-transduced with Bac-FLPo (MOI 100) and a substrate baculovirus (Bac-FCBW, MOI 100) encoding BMP-2 (a growth factor that stimulates osteogenic differentiation of ASCs), as described in Step 57C. The co-transduction enhanced and prolonged the BMP-2 expression to ∼21 d, which significantly exceeded the BMP-2 expression duration (∼7 d) conferred by a conventional baculovirus vector Bac-CB (Fig. 6)54. In parallel, the co-transduced rASCs were collected at 1 dpt, seeded to scaffolds and implanted into critical-size (8 mm in diameter) calvarial defects in NZW rabbits54. To compare the calvarial bone healing effects, two types of scaffolds were used: (i) gelatin sponge that stimulates calvarial bone healing and (ii) PLGA (poly(lactic-co-glycolic acid)) scaffold that does not promote calvarial bone healing. The co-transduced rASCs/gelatin constructs successfully filled ∼86% of the defect area and ∼61% of the defect volume at 12 weeks post transplantation, whereas the control cell/scaffold constructs (mock-transduced rASCs/gelatin and co-transduced rASCs/PLGA) failed to heal the defects (Fig. 7)54.

(a) Illustration of the BMP-2-expressing conventional baculovirus (Bac-CB) and hybrid baculovirus vectors (Bac-FCBW). (b) Time course of BMP-2 expression in rASCs. rASCs were mock-transduced, singly transduced with Bac-CB (MOI 100) or co-transduced with Bac-FLPo (MOI 50) and Bac-FCBW (MOI 100). The culture medium was collected at 1, 4, 7, 14, 21 and 28 dpt and analyzed by ELISA. The data represent the means ± s.d. of three independent experiments.

(a) Schematic illustration of experimental procedures including transduction, seeding and transplantation. rASCs were co-transduced with Bac-FLPo (MOI 100) and Bac-FCBW (MOI 100) and seeded into PLGA or gelatin sponges at 1 dpt, followed by transplantation into rabbit calvarial bone defects. (b) The maximum-intensity projection (MIP) images, microcomputed tomography (ìCT) and sagittal-view images obtained at 12 weeks post implantation. This experiment was performed on five animals, and representative data are presented showing that baculovirus-transduced rASCs/gelatin scaffolds (persistently expressing BMP-2) significantly improved calvarial bone healing in comparison with the controls (mock-transduced rASCs/gelatin and co-transduced rASCs/PLGA). Panel b is adapted from: The use of ASCs engineered to express BMP2 or TGF-âβ3 within scaffold constructs to promote calvarial bone repair, C.-Y. Lin et al., Biomaterials 34, 9401–9412, Copyright 2013, with permission from Elsevier (ref. 54).
To treat myocardial infarction (MI), rASC sheets were co-transduced with Bac-FLPo and a substrate vector expressing VEGF (a growth factor stimulating angiogenesis), as described in Step 57D. Epicardial implantation of the VEGF-expressing rASC sheet to rabbit MI models reduced the infarct size, improved cardiac functions, prevented myocardial wall thinning, suppressed myocardium fibrosis and enhanced blood vessel formation, implicating the potential of baculovirus-engineered, VEGF-expressing rASC sheet for future MI treatment22. Thus, these examples of the types of results optioned using baculovirus transduction demonstrate the potential of the hybrid baculovirus system and transduction protocol for stem cell engineering and tissue regeneration.
References
Boyce, F.M. & Bucher, N.L.R. Baculovirus-mediated gene transfer into mammalian cells. Proc. Natl. Acad. Sci. USA 93, 2348–2352 (1996).
Hofmann, C. et al. Efficient gene-transfer into human hepatocytes by baculovirus vectors. Proc. Natl. Acad. Sci. USA 92, 10099–10103 (1995).
Chen, C.-Y., Lin, C.-Y., Chen, G.-Y. & Hu, Y.-C. Baculovirus as a gene delivery vector: recent understandings of molecular alterations in transduced cells and latest applications. Biotechnol. Adv. 29, 618–631 (2011).
Hu, Y.-C. Baculovirus vectors for gene therapy. Adv. Virus. Res. 68, 287–320 (2006).
Airenne, K.J. et al. Baculovirus: an insect-derived vector for diverse gene transfer applications. Mol. Ther. 21, 739–749 (2013).
Hu, Y.-C. Baculoviral vectors for gene delivery: a review. Curr. Gene Ther. 8, 54–65 (2008).
Zeng, J. et al. Baculoviral vector-mediated transient and stable transgene expression in human embryonic stem cells. Stem Cells 25, 1055–1061 (2007).
Chen, G.-Y. et al. A graphene-based platform for induced pluripotent stem cells culture and differentiation. Biomaterials 33, 418–427 (2012).
Chen, Y.-H. et al. Baculovirus-mediated production of HDV-like particles in BHK cells using a novel oscillating bioreactor. J. Biotechnol. 118, 135–147 (2005).
Lesch, H.P. et al. Production and purification of lentiviral vectors generated in 293T suspension cells with baculoviral vectors. Gene Ther. 18, 531–538 (2011).
Kost, T.A., Condreay, J.P. & Ames, R.S. Baculovirus gene delivery: a flexible assay development tool. Curr. Gene Ther. 10, 168–173 (2010).
Liu, C.Y.-Y., Chen, H.-Z. & Chao, Y.-C. Maximizing baculovirus-mediated foreign proteins expression in mammalian cells. Curr. Gene Ther. 10, 232–241 (2010).
Grabherr, R. & Ernst, W. Baculovirus for eukaryotic protein display. Curr. Gene Ther. 10, 195–200 (2010).
Chen, C.-L. et al. Development of hybrid baculovirus vectors for artificial MicroRNA delivery and prolonged gene suppression. Biotechnol. Bioeng. 108, 2958–2967 (2011).
Hu, Y.-C., Yao, K. & Wu, T.-Y. Baculovirus as an expression and/or delivery vehicle for vaccine antigens. Expert Rev. Vaccines 7, 363–371 (2008).
Madhan, S., Prabakaran, M. & Kwang, J. Baculovirus as vaccine vectors. Curr. Gene Ther. 10, 201–213 (2010).
Wang, S. & Balasundaram, G. Potential cancer gene therapy by baculoviral transduction. Curr. Gene Ther. 10, 214–225 (2010).
Lu, C.-H. et al. Recent progresses in gene delivery-based bone tissue engineering. Biotechnol. Adv. 31, 1695–1706 (2013).
Liao, Y.-H. et al. Osteogenic differentiation of adipose-derived stem cells and calvarial defect repair using baculovirus-mediated co-expression of BMP-2 and miR-148b. Biomaterials 35, 4901–4910 (2014).
Lu, C.-H. et al. Regenerating cartilages by engineered ASCs: prolonged TGF-β3/BMP-6 expression improved articular cartilage formation and restored zonal structure. Mol. Ther. 22, 186–195 (2014).
Chen, H.-C. et al. Combination of baculovirus-mediated BMP-2 expression and rotating-shaft bioreactor culture synergistically enhances cartilage formation. Gene Ther. 15, 309–317 (2008).
Yeh, T.-S. et al. Baculovirus-transduced, VEGF-expressing adipose-derived stem cell sheet for the treatment of myocardium infarction. Biomaterials 35, 174–184 (2014).
Kost, T.A., Condreay, J.P. & Jarvis, D.L. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 23, 567–575 (2005).
Ho, Y.-C., Chen, H.-C., Wang, K.-C. & Hu, Y.-C. Highly efficient baculovirus-mediated gene transfer into rat chondrocytes. Biotechnol. Bioeng. 88, 643–651 (2004).
Wang, K.-C. et al. Baculovirus as a highly efficient gene delivery vector for the expression of hepatitis delta virus antigens in mammalian cells. Biotechnol. Bioeng. 89, 464–473 (2005).
Sung, L.-Y. et al. Modulation of chondrocyte phenotype via baculovirus-mediated growth factor expression. Biomaterials 28, 3437–3447 (2007).
Lo, W.-H. et al. Development of a hybrid baculoviral vector for sustained transgene expression. Mol. Ther. 17, 658–666 (2009).
Merrihew, R.V. et al. Chromosomal integration of transduced recombinant baculovirus DNA in mammalian cells. J. Virol. 75, 903–909 (2001).
Luo, W.-Y. et al. Adaptive immune responses elicited by baculovirus and impacts on subsequent transgene expression in vivo. J. Virol. 87, 4965–4973 (2013).
Strauss, R. et al. Baculovirus-based vaccination vectors allow for efficient induction of immune responses against Plasmodium falciparum circumsporozoite protein. Mol. Ther. 15, 193–202 (2007).
Cheshenko, N., Krougliak, N., Eisensmith, R.C. & Krougliak, V.A. A novel system for the production of fully deleted adenovirus vectors that does not require helper adenovirus. Gene Ther. 8, 846–854 (2001).
Zhou, J. & Blissard, G.W. Display of heterologous proteins on gp64 null baculovirus virions and enhanced budding mediated by a vesicular stomatitis virus G-stem construct. J Virol. 82, 1368–1377 (2008).
Hofmann, C. & Strauss, M. Baculovirus-mediated gene transfer in the presence of human serum or blood facilitated by inhibition of the complement system. Gene Ther. 5, 531–536 (1998).
Wang, C.-Y. & Wang, S. Adeno-associated virus inverted terminal repeats improve neuronal transgene expression mediated by baculoviral vectors in rat brain. Hum. Gene Ther. 16, 1219–1226 (2005).
Abe, T. et al. Involvement of the Toll-like receptor 9 signaling pathway in the induction of innate immunity by baculovirus. J. Virol. 79, 2847–2858 (2005).
Kukkonen, S.P. et al. Baculovirus capsid display: a novel tool for transduction imaging. Mol. Ther. 8, 853–862 (2003).
Condreay, J.P., Witherspoon, S.M., Clay, W.C. & Kost, T.A. Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc. Natl. Acad. Sci. USA 96, 127–132 (1999).
Hsu, C.-S., Ho, Y.-C., Wang, K.-C. & Hu, Y.-C. Investigation of optimal transduction conditions for baculovirus-mediated gene delivery into mammalian cells. Biotechnol. Bioeng. 88, 42–51 (2004).
Peng, Y., Song, J., Lu, J. & Chen, X. The histone deacetylase inhibitor sodium butyrate inhibits baculovirus-mediated transgene expression in Sf9 cells. J. Biotechnol. 131, 180–187 (2007).
Ho, Y.-C. et al. Transgene expression and differentiation of baculovirus-transduced human mesenchymal stem cells. J. Gene. Med. 7, 860–868 (2005).
Ho, Y.-C. et al. Baculovirus transduction of human mesenchymal stem cell-derived progenitor cells: variation of transgene expression with cellular differentiation states. Gene Ther. 13, 1471–1479 (2006).
Lu, L., Yu, L. & Kwang, J. Baculovirus surface-displayed hemagglutinin of H5N1 influenza virus sustains its authentic cleavage, hemagglutination activity, and antigenicity. Biochem. Biophys. Res. Commun. 358, 404–409 (2007).
Kenoutis, C. et al. Baculovirus-mediated gene delivery into mammalian cells does not alter their transcriptional and differentiating potential but is accompanied by early viral gene expression. J. Virol. 80, 4135–4146 (2006).
Shen, H.-C. et al. Baculovirus-mediated gene transfer is attenuated by sodium bicarbonate. J. Gene Med. 9, 470–478 (2007).
Sung, L.Y. et al. Enhanced and prolonged baculovirus-mediated expression by incorporating recombinase system and in cis elements: a comparative study. Nucleic Acids Res. 41, e139 (2013).
Lu, C.-H. et al. Improved chondrogenesis and engineered cartilage formation from TGF-β3-expressing adipose-derived stem cells cultured in the rotating-shaft bioreactor. Tissue Eng. Part A 18, 2114–2124 (2012).
Hu, Y.-C., Tsai, C.-T., Chang, Y.-J. & Huang, J.-H. Enhancement and prolongation of baculovirus-mediated expression in mammalian cells: focuses on strategic infection and feeding. Biotechnol. Prog. 19, 373–379 (2003).
Spenger, A. et al. Influence of promoter choice and trichostatin A treatment on expression of baculovirus delivered genes in mammalian cells. Protein Expr. Purif. 38, 17–23 (2004).
Mahonen, A.J. et al. Post-transcriptional regulatory element boosts baculovirus-mediated gene expression in vertebrate cells. J. Biotechnol. 131, 1–8 (2007).
Luo, W.Y. et al. Development of the hybrid Sleeping Beauty-baculovirus vector for sustained gene expression and cancer therapy. Gene Ther. 19, 844–851 (2012).
Lin, C.-Y. et al. The role of adipose-derived stem cells engineered with the persistently expressing hybrid baculovirus in the healing of massive bone defects. Biomaterials 32, 6505–6514 (2011).
Narsinh, K.H. et al. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 6, 78–88 (2010).
Lin, C.-Y. et al. Augmented healing of critical-size calvarial defects by baculovirus-engineered MSCs that persistently express growth factors. Biomaterials 33, 3682–3692 (2012).
Lin, C.-Y. et al. The use of ASCs engineered to express BMP2 or TGF-β3 within scaffold constructs to promote calvarial bone repair. Biomaterials 34, 9401–9412 (2013).
Turan, S. et al. Recombinase-mediated cassette exchange (RMCE): traditional concepts and current challenges. J. Mol. Biol. 407, 193–221 (2011).
Jager, L. et al. A rapid protocol for construction and production of high-capacity adenoviral vectors. Nat. Protoc. 4, 547–564 (2009).
Martinez-Morales, F. et al. Chromosomal integration of heterologous DNA in Escherichia coli with precise removal of markers and replicons used during construction. J. Bacteriol. 181, 7143–7148 (1999).
O'Reilly, D., Miller, L. & Luckow, V. Baculovirus Expression Vectors: a Laboratory Manual. (W.H. Freeman, 1992).
Papapetrou, E.P. & Sadelain, M. Generation of transgene-free human induced pluripotent stem cells with an excisable single polycistronic vector. Nat. Protoc. 6, 1251–1273 (2011).
Sugii, S., Kida, Y., Berggren, W.T. & Evans, R.M. Feeder-dependent and feeder-independent iPS cell derivation from human and mouse adipose stem cells. Nat. Protoc. 6, 346–358 (2011).
Pijlman, G.P., Van Schijndel, J.E. & Vlak, J.M. Spontaneous excision of BAC vector sequences from bacmid-derived baculovirus expression vectors upon passage in insect cells. J. Gen. Virol. 84, 2669–2678 (2003).
Zeng, J.-M. et al. High-efficiency transient transduction of human embryonic stem cell-derived neurons with baculoviral vectors. Mol. Ther. 17, 1585–1593 (2009).
Tsai, C.-T. et al. Factors influencing the production and storage of baculovirus for gene delivery: an alternative perspective from the transducing titer assay. Enzyme Microb. Technol. 40, 1345–1351 (2007).
Vannucci, L. et al. Viral vectors: a look back and ahead on gene transfer technology. New Microbiol. 36, 1–22 (2013).
Verma, I.M. & Weitzman, M.D. Gene therapy: twenty-first century medicine. Ann. Rev. Biochem. 74, 711–738 (2005).
Kay, M.A. State-of-the-art gene-based therapies: the road ahead. Nat. Rev. Genet. 12, 316–328 (2011).
Balakrishnan, B. & Jayandharan, G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 14, 1–15 (2014).
Reed, L.J. & Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27, 493–497 (1938).
Acknowledgements
We acknowledge financial support from the National Tsing Hua University (NTHU) (Toward World-Class University Project 102N2051E1, 103N2051N1); the NTHU-CGMH Joint Research Program (100N7753E1, 101N2753E1, 102N2766E1); and the Ministry of Science and Technology (NSC 101-2628-E-007-009-MY3 and NSC 101-2923-E-007-002-MY3), Taiwan. The mouse iPS cells were kindly supplied by S. Yamanaka (Center for iPS Cell Research and Application, Kyoto University).
Author information
Authors and Affiliations
Contributions
L.-Y.S., C.-L.C. and Y.-C.H. wrote the manuscript. L.-Y.S., C.-L.C., K.-C.L., C.-L.Y., S.-Y.L., G.-Y.C. and C.-Y.L. wrote individual protocols.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Sung, LY., Chen, CL., Lin, SY. et al. Efficient gene delivery into cell lines and stem cells using baculovirus. Nat Protoc 9, 1882–1899 (2014). https://doi.org/10.1038/nprot.2014.130
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2014.130
This article is cited by
-
An N-terminal conserved region in human Atg3 couples membrane curvature sensitivity to conjugase activity during autophagy
Nature Communications (2021)
-
Local magnetic activation of CRISPR
Nature Biomedical Engineering (2019)
-
Baculovirus-Mediated miR-214 Knockdown Shifts Osteoporotic ASCs Differentiation and Improves Osteoporotic Bone Defects Repair
Scientific Reports (2017)
-
Therapeutic gene editing: delivery and regulatory perspectives
Acta Pharmacologica Sinica (2017)
-
Purification of baculovirus vectors using heparin affinity chromatography
Molecular Therapy - Methods & Clinical Development (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
