
Abstract
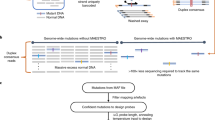
Complementary techniques that deepen information content and minimize reagent costs are required to realize the full potential of massively parallel sequencing. Here, we describe a resequencing approach that directs focus to genomic regions of high interest by combining hybridization-based purification of multi-megabase regions with sequencing on the Illumina Genome Analyzer (GA). The capture matrix is created by a microarray on which probes can be programmed as desired to target any non-repeat portion of the genome, while the method requires only a basic familiarity with microarray hybridization. We present a detailed protocol suitable for 1–2 μg of input genomic DNA and highlight key design tips in which high specificity (>65% of reads stem from enriched exons) and high sensitivity (98% targeted base pair coverage) can be achieved. We have successfully applied this to the enrichment of coding regions, in both human and mouse, ranging from 0.5 to 4 Mb in length. From genomic DNA library production to base-called sequences, this procedure takes approximately 9–10 d inclusive of array captures and one Illumina flow cell run.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


References
Kaiser, J. DNA sequencing. A plan to capture human diversity in 1000 genomes. Science 319, 395 (2008).
Siva, N. 1000 Genomes Project. Nat. Biotechnol. 26, 256 (2008).
Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Collins, F.S. & Barker, A.D. Mapping the cancer genome. Pinpointing the genes involved in cancer will help chart anew course across the complex landscape of human malignancies. Sci. Am. 296, 50–57 (2007).
Levy, S. et al. The diploid genome sequence of an individual human. PLoS Biol. 5, e254 (2007).
Wheeler, D.A. et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872–876 (2008).
Wang, J. et al. The diploid genome sequence of an Asian individual. Nature 456, 60–65 (2008).
Ley, T.J. et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456, 66–72 (2008).
Erlich, Y., Mitra, P.P., delaBastide, M., McCombie, W.R. & Hannon, G.J. Alta-Cyclic: a self-optimizing base caller for next-generation sequencing. Nat. Methods 5, 679–682 (2008).
Dohm, J.C., Lottaz, C., Borodina, T. & Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 36, e105 (2008).
Bentley, D.R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008).
Albert, T.J. et al. Direct selection of human genomic loci by microarray hybridization. Nat. Methods 4, 903–905 (2007).
Hodges, E. et al. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 39, 1522–1527 (2007).
Okou, D.T. et al. Microarray-based genomic selection for high-throughput resequencing. Nat. Methods 4, 907–909 (2007).
Porreca, G.J. et al. Multiplex amplification of large sets of human exons. Nat. Methods 4, 931–936 (2007).
Bashiardes, S. et al. Direct genomic selection. Nat. Methods 2, 63–69 (2005).
Gnirke, A. et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. (2009).
Cleary, M.A. et al. Production of complex nucleic acid libraries using highly parallel in situ oligonucleotide synthesis. Nat. Methods 1, 241–248 (2004).
Morgulis, A., Gertz, E.M., Schaffer, A.A. & Agarwala, R. WindowMasker: window-based masker for sequenced genomes. Bioinformatics 22, 134–141 (2006).
Quail, M.A. et al. A large genome center's improvements to the Illumina sequencing system. Nat. Methods 5, 1005–1010 (2008).
Lander, E.S. & Waterman, M.S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2, 231–239 (1988).
Acknowledgements
We are thankful to Danea Rebolini, Laura Cardone and Melissa Kramer for sequencing and informatic support. We also thank Mona Spector for helpful discussions. This work was supported by an NIH postdoctoral training grant (E.H.) and by kind gifts from the Stanley Foundation and Kathryn W. Davis (G.J.H.). G.J.H. is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
We declare the following conflicts of interest. Arindam Bhattacharjee, D. Benjamin Gordon, and Leonardo Brizuela are employees of Agilent, Inc. Agilent supplies arrays that can be used for the hybrid selection procedures described in this manuscript.
Rights and permissions
About this article
Cite this article
Hodges, E., Rooks, M., Xuan, Z. et al. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat Protoc 4, 960–974 (2009). https://doi.org/10.1038/nprot.2009.68
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2009.68
This article is cited by
-
Identifying the true number of specimens of the extinct blue antelope (Hippotragus leucophaeus)
Scientific Reports (2021)
-
Mycobacterium leprae diversity and population dynamics in medieval Europe from novel ancient genomes
BMC Biology (2021)
-
A seventeenth-century Mycobacterium tuberculosis genome supports a Neolithic emergence of the Mycobacterium tuberculosis complex
Genome Biology (2020)
-
The origins and adaptation of European potatoes reconstructed from historical genomes
Nature Ecology & Evolution (2019)
-
Ancient pathogen genomics as an emerging tool for infectious disease research
Nature Reviews Genetics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
