
Abstract
Complete knowledge of the genetic variation in individual human genomes is a crucial foundation for understanding the etiology of disease. Genetic variation is typically characterized by sequencing individual genomes and comparing reads to a reference. Existing methods do an excellent job of detecting variants in approximately 90% of the human genome; however, calling variants in the remaining 10% of the genome (largely low-complexity sequence and segmental duplications) is challenging. To improve variant calling, we developed a new algorithm, DISCOVAR, and examined its performance on improved, low-cost sequence data. Using a newly created reference set of variants from the finished sequence of 103 randomly chosen fosmids, we find that some standard variant call sets miss up to 25% of variants. We show that the combination of new methods and improved data increases sensitivity by several fold, with the greatest impact in challenging regions of the human genome.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others


References
1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Albers, C.A. et al. Dindel: accurate indel calls from short-read data. Genome Res. 21, 961–973 (2011).
DePristo, M.A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Gymrek, M., Golan, D., Rosset, S. & Erlich, Y. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res. 22, 1154–1162 (2012).
Iqbal, Z., Caccamo, M., Turner, I., Flicek, P. & McVean, G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 44, 226–232 (2012).
Li, B. et al. A likelihood-based framework for variant calling and de novo mutation detection in families. PLoS Genet. 8, e1002944 (2012).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
O'Fallon, B.D., Wooderchak-Donahue, W. & Crockett, D.K. A support vector machine for identification of single-nucleotide polymorphisms from next-generation sequencing data. Bioinformatics 29, 1361–1366 (2013).
Simpson, J.T. & Durbin, R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556 (2012).
Wang, Y., Lu, J., Yu, J., Gibbs, R.A. & Yu, F. An integrative variant analysis pipeline for accurate genotype/haplotype inference in population NGS data. Genome Res. 23, 833–842 (2013).
International HapMap 3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010).
Verkerk, A.J. et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991).
Lupski, J.R. et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219–232 (1991).
Kozarewa, I. et al. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Methods 6, 291–295 (2009).
Morgulis, A., Gertz, E.M., Schaffer, A.A. & Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 13, 1028–1040 (2006).
She, X. et al. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature 431, 927–930 (2004).
Ross, M.G. et al. Characterizing and measuring bias in sequence data. Genome Biol. 14, R51 (2013).
Li, H. Improving SNP discovery by base alignment quality. Bioinformatics 27, 1157–1158 (2011).
Gatchel, J.R. & Zoghbi, H.Y. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 6, 743–755 (2005).
Labeit, S., Ottenheijm, C.A. & Granzier, H. Nebulin, a major player in muscle health and disease. FASEB J. 25, 822–829 (2011).
Efron, B. Bootstrap methods: another look at the jackknife. Ann. Stat. 7, 1–26 (1979).
Chin, C.S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Otto, T.D., Sanders, M., Berriman, M. & Newbold, C. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 26, 1704–1707 (2010).
Chain, P.S. et al. Genome project standards in a new era of sequencing. Science 326, 236–237 (2009).
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Myers, E.W. et al. A whole-genome assembly of Drosophila. Science 287, 2196–2204 (2000).
Venter, J.C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Pevzner, P.A., Tang, H. & Waterman, M.S. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA 98, 9748–9753 (2001).
Zerbino, D.R., McEwen, G.K., Margulies, E.H. & Birney, E. Pebble and Rock Band: heuristic resolution of repeats and scaffolding in the Velvet short-read de novo assembler. PLoS ONE 4, e8407 (2009).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 108, 1513–1518 (2011).
Batzoglou, S. et al. ARACHNE: a whole-genome shotgun assembler. Genome Res. 12, 177–189 (2002).
Kao, W.C., Chan, A.H. & Song, Y.S. ECHO: a reference-free short-read error correction algorithm. Genome Res. 21, 1181–1192 (2011).
Butler, J. et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820 (2008).
Acknowledgements
We thank the Broad Institute Genomics Platform for generating data for this project and J. Bochicchio for project management. We thank the 1000 Genomes Project for early access to 250-base reads from the Centre d′Etude du Polymorphisme Humain (CEPH) trio. We thank Illumina for the use of sequence data from the 17-member CEPH pedigree and M. Eberle for detailed information on these data. We thank Z. Iqbal for detailed advice regarding running Cortex on 250-base reads and M. Carneiro and E. Banks for detailed advice regarding running GATK on the same data. We thank H. Li for early access to bwa-mem and for his extensive examination of our manuscript. We thank S. Fisher for help preparing the data cost estimate. We thank M. Daly, M. DePristo, S. Gabriel, Z. Iqbal, D. MacArthur, D. Neafsey and M. Zody for helpful comments. This project has been funded in part with federal funds from the National Human Genome Research Institute, US National Institutes of Health, US Department of Health and Human Services, under grants R01HG003474 and U54HG003067, and in part with federal funds from the National Institute of Allergy and Infectious Diseases, US National Institutes of Health, US Department of Health and Human Services, under contract HHSN272200900018C.
Author information
Authors and Affiliations
Contributions
D.B.J., I.M. and C.R. designed and managed the project. S.Y., N.I.W., T.S., B.L., I.M. and D.B.J. designed algorithms and analyzed data. R.H., L.H., B.S., D.T. and L.W. designed and executed laboratory processes. D.B.J., C.N. and E.S.L. shaped the results and developed the manuscript, to which all authors contributed. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Nearby mutations involving indels not deducible from single-sample DNA.
(a,b) The same sample DNA is aligned to the same reference sequence in two different ways. In both a and b, the DNA appears to represent a deletion and a substitution in the sample, but the mutations are different. Given data from enough samples, one might infer the actual biological history of this complex event.
Supplementary Figure 2 DISCOVAR assembly of segmental duplication in the nebulin gene.
The gene for nebulin (NEB) contains three tandem and highly similar copies of a 10-kb repeat. We ran DISCOVAR on the NA12878 data set of this work, using hg19 region chr. 2: 152,433,184–152,468,843. The figure exhibits the assembly graph. Each edge represents a DNA sequence and is labeled by a numerical identifier (corresponding to a record in a FASTA file; not shown). Edges are color-coded by length, with gray representing the shortest sequences and increasing lengths represented by black, red and finally magenta (none shown). The circular part (loop) is traversed three times, entering at edge 52 and exiting at edge 1. Edges 41 and 23 would be seen to continue on were the boundaries of the region to be extended. The number of loop traversals (3) can be determined by coverage and is consistent with the reference sequence, although a different allelic balance than 3 + 3, e.g., 2 + 4, cannot be absolutely ruled out. Each edge either aligns perfectly to one of the three repeat copies or else represents a difference with one (or more) of them. Some edges carry variants that may be assigned to particular repeat copies, whereas other edges carry variants for which the repeat copy cannot be determined. For example, edge 6 aligns perfectly to the first two repeat copies and with six mismatches to the third copy. Because the graph topology implies that each repeat copy passes through edge 6, it follows that in fact six variants can be inferred for the third repeat copy. (This might be explained by gene conversion.) Other changes (for example on edge 4) cannot be localized to the repeat copy.
Supplementary Figure 3 Fragment size distribution for Illumina 250-base read pairs.
For the data set of this work, read pairs were aligned to the hg19 reference and the distribution of fragment sizes was inferred from this. The x axis shows the fragment size in basepairs, and the y axis shows the number of fragments observed for each size.
Supplementary Figure 4 Error correction and read pair closure.
(a) For each read, termed the ‘founder’, all reads sharing a K-base perfect match with it are found. These are the founder’s ‘friends’ and define a ‘stack’ of reads. In general, different friends have different matches with the founder. (b) Some friends are declared false and are removed from the stack. In particular, if there is a column in which the friend and a founder are assigned high quality scores but have different base calls (illustrated as red and green), the friends having these discrepant bases (shown as gray) are declared false. (c) Given a read pair (two thick black segments), the friends of both are found, the right stack is reverse complemented and their stacks are then truncated on the left and right. Then, the consensus sequences (red and blue) of the left and right stacks are found. By aligning the consensus sequences to each other, the relative positions of the two stacks may be determined. The two stacks are then merged, yielding a ‘joint stack’. The consensus of the joint stack defines the read pair closure (green).
Supplementary Figure 5 Graph reconstruction.
(a) Left, an assembly having five edges, x1, x2, r, y1 and y2, is depicted. The two red paths represent read pair closures x1ry1 and x2ry2. This is what one would likely obtain if r were sufficiently short. The assembly graph is reconstructed by gluing the closure paths together along all proper (end-to-end) overlaps (none in this case). Right, the assembly graph reconstruction arising from this gluing. We suppose that this is the exact genome structure. (b) Left, four paths are shown, as might arise if r were sufficiently long. Right, the reconstruction recapitulates the initial assembly graph. (c) Left, six paths are shown, as would be expected for r of intermediate length. The assembly graph is again reconstructed by gluing along all proper overlaps. Right, the initial assembly graph is again obtained. (d) Left, the same six paths are shown, but we suppose that certain overlaps x1r/ry2 and x2r/ry1 are somehow prohibited. Right, with this restriction, after gluing the genome is obtained. (e) Hypothetical data, showing closure paths (left three columns) and their multiplicities within the data (rightmost column), that might lead us to conclude that overlaps x1r/ry2 and x2r/ry1 should be prohibited. The given matrix is for all closure paths that contain the edge r. Examining the matrix, we observe, for example, x1r and ry2 but not x1ry2. Moreover, there are many paths that traverse r, indicating ample coverage in the region, and every row in the matrix is explained by either x1ry1 or x2ry2. This allows us to prohibit x1r/ry2, and similarly x2r/ry1.
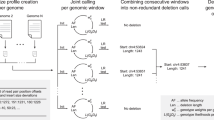
Supplementary Figure 6 Variant calling.
A condensed view of the variant calling algorithm (Supplementary Note, Section 6) is shown. (a) The assembly graph. (b) Edges are shown in alignment to the reference sequence. A given edge can appear in more than one location. (c) An acyclic graph is constructed from the data in b. (d) Variants are deduced from the graph.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6, Supplementary Tables 1–10 and Supplementary Note. (PDF 1190 kb)
Variants called by each caller in fosmid regions.
This data set provides a tab-delimited list of the variants called in the fosmid regions showing the fosmid identifier, position, reference allele alternate allele and a comma-separated list of notes. The notes are either “fosmid,” indicating that the variant is present in the fosmid reference set or of the form caller=state, where caller is Platinum-100, GATK-250, DISCOVAR or Cortex and state is het, hom or hom+. Here het means that both the variant and reference were called, hom means that the variant but not the reference was called and hom+ means that the variant was called as homozygous. This data set was automatically generated. See also the manually identified defects noted in Supplementary Table 1a. (TXT 442 kb)
Rights and permissions
About this article

Cite this article
Weisenfeld, N., Yin, S., Sharpe, T. et al. Comprehensive variation discovery in single human genomes. Nat Genet 46, 1350–1355 (2014). https://doi.org/10.1038/ng.3121
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3121
This article is cited by
-
Immune gene variation associated with chromosome-scale differences among individual zebrafish genomes
Scientific Reports (2023)
-
Whole genome assembly of the armored loricariid catfish Ancistrus triradiatus highlights herbivory signatures
Molecular Genetics and Genomics (2022)
-
Variation in the expression of a transmembrane protein influences cell growth in Arabidopsis thaliana petals by altering auxin responses
BMC Plant Biology (2020)
-
LRScaf: improving draft genomes using long noisy reads
BMC Genomics (2019)
-
Linear Mitochondrial Genome in Anthozoa (Cnidaria): A Case Study in Ceriantharia
Scientific Reports (2019)
