
Abstract
The woodland strawberry, Fragaria vesca (2n = 2x = 14), is a versatile experimental plant system. This diminutive herbaceous perennial has a small genome (240 Mb), is amenable to genetic transformation and shares substantial sequence identity with the cultivated strawberry (Fragaria × ananassa) and other economically important rosaceous plants. Here we report the draft F. vesca genome, which was sequenced to ×39 coverage using second-generation technology, assembled de novo and then anchored to the genetic linkage map into seven pseudochromosomes. This diploid strawberry sequence lacks the large genome duplications seen in other rosids. Gene prediction modeling identified 34,809 genes, with most being supported by transcriptome mapping. Genes critical to valuable horticultural traits including flavor, nutritional value and flowering time were identified. Macrosyntenic relationships between Fragaria and Prunus predict a hypothetical ancestral Rosaceae genome that had nine chromosomes. New phylogenetic analysis of 154 protein-coding genes suggests that assignment of Populus to Malvidae, rather than Fabidae, is warranted.
Similar content being viewed by others

Main
The cultivated strawberry, F. × ananassa, originated ∼250 years ago and is among the youngest crop species1. Botanically, it is neither a berry nor a true fruit, as the actual fruit consists of the abundant dry achenes (or seeds) that dot the surface of a fleshy modified shoot tip, the receptacle. Unlike other Rosaceae family crops such as apple and peach, the strawberry is considered to be non-climacteric because the flesh does not ripen in response to ethylene. Genomically, F. × ananassa is among the most complex of crop plants, harboring eight sets of chromosomes (2n = 8x = 56) derived from as many as four different diploid ancestors. Paradoxically, the small basic (x = 7) genome size of the strawberry genus, ∼240 Mb, offers substantial advantages for genomic research.
An international consortium selected F. vesca (2n = 2x = 14) for sequencing as a genomic reference for the genus2. The so-called 'semperflorens' or 'alpine' forms of F. vesca ssp. vesca have been cultivated for centuries in European gardens1. Their widespread temperate growing range, self-compatibility and long history of cultivation, coupled with selection for favorable recessive traits such as day neutrality, non-runnering and yellow-fruited forms offer extensive genotypic diversity. More broadly, F. vesca offers many advantages as a reference genomic system for Rosaceae, including a short generation time for a perennial, ease of vegetative propagation and small herbaceous stature compared with tree species such as peach or apple. Robust in vitro regeneration and transformation systems have been established for F. vesca, facilitating the production of forward and reverse genetic tools as well as structural and functional studies3,4,5,6. These properties render strawberry an attractive surrogate for testing gene function for all plants in the Rosaceae family.
This report presents the genome sequence of the diploid strawberry F. vesca ssp. vesca accession Hawaii 4 (National Clonal Germplasm Repository accession # PI551572). We achieved coverage exclusively with short-read technologies and did assembly without a physical reference, demonstrating that a contiguous plant genome sequence can be assembled and characterized using solely these technologies. Moreover, this genome was sequenced using an open-access community model.
Results
Genome sequencing and assembly
We selected a fourth-generation inbred line of the F. vesca ssp. vesca accession Hawaii 4 known as 'H4×4' for sequencing primarily because of its amenability to high-throughput genetic transformation. The Hawaii 4 accession was used for transfer DNA (T-DNA) insertional mutagenesis5,6, as well as transposon and activation tagging. H4x4 is day neutral, sets abundant seeds on self-pollination and completes a life cycle in 4–6 months regardless of season. It has white-yellow fruit and produces new plants from modified stems called stolons.
We used the Roche/454, Illumina/Solexa and Life Technologies/SOLiD platforms to generate ×39 combined average coverage (Online Methods). A summary of the input sequence data used for the assembly is presented in Supplementary Table 1. Over 3,200 scaffolds were assembled with an N50 of 1.3 Mb (Supplementary Table 2). Over 95% (209.8 Mb) of the total sequence is represented in 272 scaffolds. Resequencing using Illumina confirmed the high quality of the assembly, with 99.8% of the scaffolds and 99.98% of the bases in the assembly being validated by perfect-match Illumina reads with an average depth of approximately ×26 (Supplementary Fig. 1). The F. vesca H4x4 genome size was estimated at ∼240 Mb using flow cytometry, with Arabidopsis thaliana (∼147 Mb) and Brachypodium distachyon (300 Mb) serving as internal controls (Supplementary Table 3).
Anchoring genome sequence to the genetic map
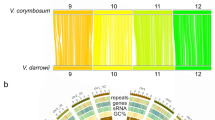
We aligned and oriented the scaffolds of the assembly to the diploid Fragaria reference linkage map (FV × FN) and its associated bin map7 (Fig. 1). Of 272 F. vesca H4x4 sequence scaffolds that were composed of over 10,000 bp (a total of 209.8 Mb of scaffold sequences and embedded gaps), 131 were anchored to the FV × FN map. The scaffolds were anchored by 320 genetic markers, including 234 mapped in the full FV × FN progeny6 (Fig. 1, blue bars) and 86 mapped in a bin set of six seedlings7 (Fig. 1, yellow bars). Additionally, we used a new method to identify segregating SNP markers through direct Illumina sequencing of a reduced complexity AluI digestion of the bin set seedling DNA for anchoring 70 additional scaffolds of over 100,000 kb in length to the genetic map. Three scaffolds mapped to two locations on the genetic map and were split into two at regions of low coverage. Thus, a total of 204 genome sequence scaffolds (including the three split scaffolds) containing 198.1 Mb of sequence data (∼94% of the total scaffold sequence) was anchored to the FV × FN map using 390 markers. We assembled the scaffolds into seven pseudochromosomes, numbered according to the linkage group nomenclature used in a previous study8.

Scaffolds representing 198.1 Mb of scaffolded sequence with embedded gaps (99.2% of all contiguous sequence over 10 kb in length) were anchored to the genetic map with 390 genetic markers. Blue scaffolds were anchored and oriented using map positions of markers in the full FV × FN progeny, whereas the yellow scaffolds were anchored to mapping bins.
Although a comprehensive molecular karyotype has yet to be established for F. vesca, researchers in a previous study9 identified, by fluorescent in situ hybridization (FISH), three pairs of 45S (18S-5.8S-25S) loci and one pair of 5S loci that co-localized with one of the pairs of 45S loci in an unspecified accession of F. vesca. We also found these karyotypic features in F. vesca H4x4 and identified tentative correspondences between two cytologically marked chromosomes and genomically defined pseudochromosomes using mitotic (root tip) chromosomes hybridized to 25S (red) and 5S (green) rDNA probes (Online Methods and Supplementary Fig. 2). Chromosome G displayed a strong distal 25S signal and a proximal 5S signal, whereas chromosome F displayed a strong distal 25S signal and chromosome D displayed a weak distal 25S signal. The 5S probe sequence had sequence homology to two small scaffolds that are not mapped to pseudochromosomes and one scaffold that maps to pseudochromosome VII at the locus defined by marker EMFv190. The 25S sequence had 32 matches of >90% sequence identity, of which 30 were unmapped scaffolds of less than 1.7 kb. Pseudochromosome VII, with mapped scaffolds containing 25S and 5S sequences at distal and proximal locations, respectively, appears to correspond to chromosome G. Pseudochromosome VI, which also contains 25S sequences in a mapped scaffold may correspond to chromosome F. No mapped scaffold could be implicated as corresponding to the weak 25S signal on chromosome D.
Synteny infers ancestral relationships
Genome-wide analyses provide insight into the nature and dynamics of macro-syntenic relationships among rosaceous taxa. Comparison of the map positions of 389 rosaceous conserved ortholog set (RosCOS) markers previously bin mapped in Prunus10 to their positions on the seven pseudochromosomes of F. vesca H4x4 revealed macro-syntenic relationships between the two genomes (Fig. 2). Markers were deemed orthologous between the two genomes when five or more RosCOS occurred within 'syntenic blocks' shared between the two genomes. This analysis revealed remarkable genome conservation between the two taxa, with complete synteny between Prunus linkage group (PG) 2 and Fragaria chromosome (FC) 7, PG8 and a section of FC2, and PG5 with a section of FC5. Most markers mapped to PG3 were located on FC6, with the remainder being on FC1. Markers on PG4 located to FC3 and FC2, whereas those on PG7 mapped onto FC6 and FC1. New chromosomal translocations between PG1-FC5, PG3-FC1 and PG6-FC6 were identified, adding improved resolution to a previous study7. Our data support these same broad structural relationships, providing extensive evidence for the reconstruction of an ancestral genome for Rosaceae with a haploid chromosome number (x) of nine, consistent with the base haploid chromosome number of the largest group within the modern Rosaceae, the Spiraeoideae11.

The diagram was plotted using Circos; map positions from the Prunus reference map were converted to approximate physical positions for comparison by multiplying the marker positions in cM by 400,000. Markers were spaced at 100,000 nucleotide intervals within each T × E mapping bin (see URLs).
Absence of large duplications in the F. vesca genome
A comparison of the F. vesca genome against itself using MUMmer12 version 3.22 (Online Methods) showed that long matches form eight distinct families of approximate repeats, with the largest family having 15 occurrences (Supplementary Fig. 3). This family shows homology to a rice retrotransposon protein (GenBank: ABA95102.1)13 and contains the longest (14,721 bp) of the 126 matches that are ≥10,000 bp. All other families have three occurrences or less. F. vesca is the only plant genome sequenced to date with no evidence of large-scale, within-genome duplication (Supplementary Fig. 3). All members of the rosid clade share an ancient triplication, first documented in grape14 and found in all other rosid genomes, including apple15. In strawberry, chromosome rearrangement (Fig. 2) and genome size reduction (perhaps accompanied by preferential loss of duplicated genes) may obscure the signature of the ancient triplication.
Repetitive sequences and transposable elements
In all plants studied, transposable elements are major components of genomes, both in the percentage of the nuclear genome they represent and the degree to which they drive gene and/or genome evolution. Extensive homology and structure-based searches of the F. vesca genome, performed as previously described for the much larger maize genome16, identified 576 different transposable element exemplars17, including more than 6,000 fully intact transposable elements (Supplementary Table 4). These elements mask about 22% of the assembly, compared to ∼1.3% of the assembly masked by transposable elements in Repbase18, the Munich Information Center for Protein Sequences (MIPs) repeat database19 and the Institute for Genomic Research (TIGR) repeat database20 combined. No statistically significant difference was found in the amount of data masked in the raw sequence reads compared to the assembly, indicating that the assembly provides a comprehensive transposable element discovery resource. Moreover, previously identified transposable elements21 and nine intact long terminal repeat (LTR) retrotransposons of the Copia superfamily were identified by the structure-based and homology-based searches of 30 sequenced F. vesca spp. americana fosmids22.
LTR retrotransposons occupy ∼16% of the F. vesca nuclear genome, whereas CACTA elements and miniature inverted-repeat transposable elements (MITEs), the most numerous DNA transposable elements, represent 2.8% and 2.4%, respectively, of nuclear DNA. The most numerous LTR retrotransposon family has fewer than 2,100 copies. Average-size angiosperm genomes have families with copy numbers greater than 10,000, so the lack of highly abundant LTR retrotransposons is likely to be the reason F. vesca has a relatively small-size genome. High sequence identity between some elements suggests recent transposition activity.
Transcriptome sequence
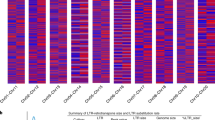
Multiple complex complementary DNA (cDNA) pools provided F. vesca transcriptome sequence resources for gene model prediction and validation23 and highlighted organ specificity of fruit and root transcripts. Analysis of the relative representation of genes in the fruit- and root-specific cDNA libraries (Online Methods) identified transcripts of overrepresented genes (>twofold, false discovery rate < 0.01) in the fruit (1,753 genes) and root (2,151 genes). A global perspective of the expression patterns of these genes in roots and fruit is provided (Fig. 3). Genes overrepresented in fruit showed enrichment for several categories of biological processes and molecular functions associated with fruit development and were dominated by genes related to carbohydrate metabolic activity (glycosyltransferases, pectin esterases and polygalacturonases). Flower, fruit and embryo development, maturation-associated amino acid metabolism, secondary metabolic and lipid metabolic genes were also overrepresented, consistent with other reports24. In contrast, abundant transcripts in roots represented transcription factor, kinase and signal transduction categories. We observed overrepresentation of biotic and abiotic stress-related genes in roots compared to fruit. Results confirmed the expected transcriptional plasticity between different organs and developmental stages.

Overrepresented gene ontology categories in fruit (a) and root (b) expressed genes. The circles are shaded based on significance level (yellow, false discovery rate < 0.05), and the radius of each circle denotes the number of genes in each category.
Gene prediction and models
We implemented a new machine learning algorithm, GeneMark-ES+, which combines ab initio gene predictions, evidence for host gene deserts from the F. vesca transposable element library and external evidence for gene elements derived from the transcriptome sequencing, to optimize precision of F. vesca gene annotation (Supplementary Fig. 4). Parameters of the ab initio gene finder GeneMark-ES25 were defined by unsupervised training on the whole unmasked sequence of F. vesca genome; hybrid gene models were generated for the genomic sequence masked for repetitive elements and marked for introns inferred from a high confidence set of F. vesca transcript sequences. This process generated 34,809 hybrid gene models with a mean coding sequence size of 1,160 nt and a mean of 4.8 exons per gene (Supplementary Table 5). Predicted protein sequences were searched for similarity by BLAST against SwissProt, UniRef90, RefSeq (plant) databases and A. thaliana proteins (Supplementary Table 6). At least one Interpro motif26 was detected in 63% of hybrid gene models. Based on our functional annotation pipeline, we provided preliminary annotation for approximately 25,050 genes (Supplementary Fig. 5a,b). Gene clustering methods allowed us to computationally assign gene families to 18,170 genes (∼55%).
Comparison of transcriptome sequence information to gene models using the Illumina RNA-seq and Roche/454 EST sequences provided empirical support for the predictions (Supplementary Figs. 6a and 6b). Our approach to gene prediction proved highly effective as 90% of hybrid gene models were supported by transcript-based evidence. These findings also confirm the completeness of genome sequence coverage. Gene models, transcriptional support and analytical tools may be accessed through the strawberry genome browser (see URLs).
RNA genes
We identified RNA genes in the assembly: 569 transfer RNAs (tRNAs), 177 ribosomal RNA (rRNA) fragments, 111 spliceosomal RNAs, 168 small nucleolar RNAs, 76 micro RNAs and 24 other RNAs (Supplementary Table 7). These include the minor ATAC spliceosomal RNAs and the thiamin pyrophosphate riboswitch that controls alternative splicing of THIC mRNA27. Although organellar sequences were generally underrepresented in the assembly, we did recover several organellar RNA sequences. We found no full-length copies of large cytoplasmic rRNAs; however, there was sufficient coverage along the length of large rRNAs to produce consensus sequences (Supplementary Table 8).
Chloroplast genome
The H4x4 chloroplast genome is 155,691 bp long and encodes 78 proteins, 30 tRNAs and four rRNA genes. Noteworthy is the absence of the atpF group II intron. This absence has previously not been found in land plant chloroplast genomes outside of Malpighiales28. We also observed evidence for recent DNA transfer from the plastid to the nuclear genome (chloroplast nomads) (Supplementary Fig. 7). The correlation of reduced sequence identity with shorter inserts is similar to the pattern reported in Sorghum29.
Gene ontology annotation
Annotation coverage in the strawberry genome is equivalent to that of Arabidopsis, which has a genome of similar size. Preliminary annotation of ∼25,050 genes (Supplementary Fig. 5a,b) suggested that the F. vesca genome maintains more genes for 'molecular function' categories defined for transport, signal transduction and structural molecules. Roughly the same number of genes was assigned to catalytic activity, whereas more were assigned for biological processes, such as transport, protein metabolism and response to freezing. Additional gene counts with cellular localization to the mitochondria, plastid, membrane, ribosome, cytoskeleton and chromosome might be due to the enriched gene ontology annotation methods employed in this study.
Multiple genome alignment to F. vesca as anchor
Eight plant genomes were aligned, anchored by the genome of F. vesca (Online Methods). The other seven plants represent the most closely related available genomic sequences. Supplementary Table 9 (for the complete version of this table, see link in the URL section) shows that Vitis vinifera and Populus trichocarpa share the most genes with F. vesca. This genome alignment is independent of gene predictions, as it is based on translated nucleotide sequences; 87% of the conserved regions overlap coding sequences from gene predictions, thus providing evidence that the CDSs and overlapping conserved regions are indeed true coding sequences.
Strawberry unique gene clusters
A total of 103,570 gene sequences from a monocot (rice) and three dicots (Arabidopsis, grape and strawberry) clustered together in 15,969 gene families (Fig. 4). Of the 33,264 protein-coding genes in strawberry, 18,170 genes aligned in 9,895 of these gene family clusters, with 681 gene clusters being unique to strawberry. These 681 gene clusters represent 957 genes, of which 416 contain InterPro domains and were assigned gene ontology categories. The remaining 541 are previously unidentified predicted genes of unknown function. These numbers are consistent with relative proportions from other sequenced genomes. The most InterPro domains found belong to transcription factor categories, followed by kinase domains and enzymatic activities related to fruit development, ripening and sugar metabolism. Of the 1,753 genes overrepresented in the fruit transcriptome, 92 belong to the strawberry-unique clusters and 84 of these are previously unidentified genes with no known InterPro or gene ontology classification. Similarly, of the 2,151 genes overrepresented in the root transcriptome, 133 belong to the strawberry-specific category, of which 128 are previously unidentified genes with no known InterPro or GO assignments.

Comparative analysis with rice, Arabidopsis, grape and strawberry genes revealed that a total of 103,570 genes from those four species were shared among all four species. In the case of strawberry, 18,170 genes of the total 33,264 protein-coding genes (from ab initio predictions; Supplementary Table 5) aligned in 9,895 clusters. Comparison of the four species revealed 681 gene clusters unique to strawberry. There were 663 gene clusters unique to strawberry and Arabidopsis, whereas there were 262 gene clusters unique to rice and strawberry. Additionally, there were 6,233 gene clusters that were shared among all four species. The analysis was done using a total of 21 species to find the clusters.
By comparison, 2,777 clusters were unique to the monocot. Approximately the same number of clusters were shared by the well-annotated eudicot and monocot models, Arabidopsis and rice (260), as are shared by the two perennial fruit crops, grape and strawberry (269), although 6,233 gene clusters were common to all four species. These data represent a subset of the analysis of gene sequences from plant and non-plant species that represent a uniform distribution across the tree of life and have completely sequenced genomes with annotated genes.
An opportunity for translational studies
Studies in model species such as A. thaliana have defined basic tenets of plant biology. The diploid strawberry represents a parallel system for testing these paradigms in an agile translational system. The F. vesca sequence permits access to genomic information relevant to Rosaceae, especially fruit quality (flavor, nutrition and aroma). However, only about 100 genes central to these processes have been functionally characterized in Fragaria (Supplementary Tables 10 and 11). Analysis of the F. vesca genome has revealed orthologs and paralogs of many structural genes (Supplementary Tables 11, 12, 13, 14, 15 and 16) involved in key biological processes such as flavor production, flowering and response to disease (Supplementary Note). Flavors and aromas arise from the perception of volatile compounds mainly produced by the fatty acid, terpenoid and phenylpropanoid metabolic pathways. Several gene families have been implicated in the production of these volatile components, including the acyltransferases, the terpene synthases and the small molecule O-methyltransferases (Supplementary Table 11 and Supplementary Fig. 8). Examination of the strawberry genome revealed an intact flowering molecular circuit that parallels Arabidopsis and encompasses genes controlling the sensing of light (cryptochromes and phytochromes) through the circadian oscillator (Supplementary Table 12 and Supplementary Fig. 9). Genes controlling the production of jasmonic acid (Supplementary Table 13), salicylic acid (Supplementary Table 14), nitric oxide (Supplementary Table 15) and pathogenesis-related proteins (Supplementary Table 16) have been associated with disease resistance in various species30,31, indicating that a core set of signal transduction elements is shared between strawberry and other plants.
Analysis of transcription factor families
Within the F. vesca genome, we identified 1,616 transcription factors (Supplementary Table 17), compared to 1,403 for grape and 1,856 for Arabidopsis using the same stringent BLAST-identity. MYB transcription factors have been implicated in regulating diverse plant responses, including growth or regulation of primary (sucrose) and secondary (lignins and phenylpropanoids) metabolites in response to hormones, abiotic and biotic stress, and light and circadian rhythm. Overall, Arabidopsis has 303 MYB and MYB-related transcription factors, whereas F. vesca has 187. Phylogeny of the R2R3 MYBs (Supplementary Fig. 10) showed orthologs of many Arabidopsis genes with assigned function, for example, those that underlie gene expression relevant to the production of flavonols and proanthocyanins.
BLAST analysis of 20 Arabidopsis R2R3 MYB sequences representing transcription factors implicated in regulating the phenylpropanoid metabolic pathway against the F. vesca genome identified 25 highly homologous sequences (Supplementary Table 18). The greatest clade expansion was around TT2 (encoding transparent Testa 2, also known as MYB123), which controls proanthocyanidin levels in the seed32. There are at least six strawberry TT2-like MYBs. However, when Illumina-sequenced cDNA read mapping of these genes was considered, two appeared to be silent, five were expressed, and one (FvMyb33; gene08694) was highly expressed, suggesting a key function in strawberry proanthocyanidin synthesis. This MYB gene has been duplicated in Brassica napus and Lotus japonicus32,33.
Reinterpretation of angiosperm phylogeny
Comparative analysis with other angiosperm genomes surprisingly revealed that the currently accepted phylogenetic placement of Populus, an important model organism for tree crops, may be incorrect. Gene selection began with an earlier gene tree database34 of 9,000 homology groups, searching for genes that were single copy, and filtering by a measure of phylogenetic coherence (Online Methods and Supplementary Note), which yielded 240 genes from seven species. Upon addition of homologs from newly sequenced genomes (Glycine, Carica and Lotus), complex duplication patterns required rejecting some genes, whereas simple terminal duplications were resolved by selecting a single product. This yielded 154 genes present in at least eight of the ten species totaling 68,526 aligned amino acid positions. Maximum likelihood phylogenetic analysis produced a tree (Fig. 5) that differed from most earlier studies (discussed below) in placing Populus with Arabidopsis in Malvidae rather than with Fragaria and legumes in Fabidae.

The tree is based on alignments of 154 genes present in at least eight of ten genomes. Genes exhibiting little or no duplication were selected, and duplicates, predominant in Glycine, were removed. Species in the Fabidae clade are colored red and species in the Malvidae clade are colored blue. The placement of Populus in Malvidae and not Fabidae, as found in previous studies, was strongly supported by topology and resampling tests. Bootstrap values are shown at nodes. The scale is amino acid substitutions per site.
The approximately unbiased test35 using the 154-gene set rejected the monophyly of Fabidae with a P value of 3 × 10−38. Analysis of the subset of 87 genes present in nine or more species (38,840 AA positions) and the subset of 24 genes present in all ten species (9,480 AA positions) yielded the same topology. Resampling genes from the 154- and 87-gene sets (50% genewise jackknifing36) fully supported all clades except Medicago-Lotus (98%), demonstrating robustness of gene choice. We used bootstrapping the per-site likelihoods for the best tree versus the Fabidae topology to determine the minimum number of positions needed to reduce support for the Fabidae topology to less than 1% of replicates. The minimum was 5,120 positions (55%) for 24 genes, 13,594 positions (35%) for 87 genes and 13,704 positions (20%) for 154 genes. This shows a decline in resolving power per position as we included more non-universal genes, though the larger datasets accumulated more overall power.
Phylogenetic analyses of angiosperms based exclusively on chloroplast genes have consistently resolved with strong support two large rosid clades, Fabidae and Malvidae37. In contrast, studies based on the mitochondrial gene matR38 and 13 protein-coding genes39, as well as four plastid, six mitochondrial, and three nuclear genes40, placed Populus (and its order, Malpighiales) in Malvidae. However, this topology had either <50% maximum likelihood bootstrap support38,39, or taxonomic sampling of Malvidae was limited to Arabidopsis40. A recent phylogenetic analysis of four mitochondrial genes obtained strong support for the non-monophyly of Fabidae41. Together with our nuclear gene results, there are now two independent sources of evidence for placing Populus in Malvidae and not Fabidae. Consistent with this result, there are at least seven floral characters42 that suggest the Malpighiales share a common ancestor with Malvidae and no shared derived characters for Fabidae, including Malpighiales.
These apparently conflicting results may be due to biological differences between chloroplast and nuclear evolution. Chloroplasts lack the history of frequent gene duplications and loss that can lead to errors in recognizing orthologs in nuclear genes. Chloroplasts, however, can experience inheritance at odds with the genome as a whole43, especially when speciation events are compressed in time, as hypothesized for the rapid radiation of the rosid orders 83–108 million years ago37. As more plant genomes are sequenced, these apparent discrepancies will be clarified by combining the advantages of copious genomic data44 with denser taxonomic sampling.
Discussion
The genome sequence for F. vesca is the smallest sequenced plant genome other than Arabidopsis and represents a gateway to functional gene studies within Rosaceae. Like Arabidopsis, F. vesca is rapidly transformable, grows with a small footprint and has a short generation time from seed to seed, which are all traits that make it particularly useful for functional genomics research. Unlike Arabidopsis, F. vesca is perennial. Its nearest relatives are high-value fruit crops with cumbersome polyploid genomes, such as cultivated strawberry, or large statured crops with long generation times and/or spatial requirements, such as apple, rose, cherry or peach. Economically important traits including disease resistance, developmental controls and fruit flavor and quality can be addressed with this agile system. Completion of the sequencing of the strawberry genome also illustrates that a plant genome can be sequenced and assembled using exclusively short-read technology without a physical map or reference genome.
URLs.
Strawberry genome browser, http://www.strawberrygenome.org; the complete version of Supplementary Table 9, http://staff.vbi.vt.edu/setubal/mapG.html; HashMatch, http://mocklerlab-tools.cgrb.oregonstate.edu/; Circos, http://mkweb.bcgsc.ca/circos/.
Methods
Genome sequencing and assembly.
The assembly of the strawberry genome was accomplished using Celera Assembler (CA) version 5.3 as the primary assembly engine. Input data are summarized in Supplementary Table 1 1. For 454 reads, values in the rightmost three columns represent what remained after trimming reads, having removed those below the minimum length threshold for CA (64 bp) and removing duplicate reads and mate pairs. For Illumina reads, values in the right columns represent what remained after removing reads containing either ambiguous base calls or adaptor sequence. For SOLiD reads, the right columns represent the number of pairs where each end could be uniquely mapped to assembly contigs, with redundant pairs removed.
All 454 input data were converted from the native .sff file format to CA.frg file format. Linker sequences remaining in reads, after detecting mates, were found using NUCmer, and fragment clear ranges in input files were modified to exclude them. The CA pipeline was then run on a single large-memory multiprocessor computer (128 Gb memory and 32 processors) with default parameters, except for parameters to run multiple processes on a single computer, resulting in an initial set of contigs.
The paired-end SOLiD reads were then mapped to these contigs using Bowtie45 to create additional mate pairs that were added to CA inputs, and the scaffolding step was run to create a set of scaffolds. A separate assembly of the Illumina/Solexa reads was done with Velvet version 0.7.3146. The resulting Velvet contigs were mapped to existing scaffolds using NUCmer47. Where a Velvet contig mapped unambiguously to contig sequences flanking a scaffold gap, the corresponding Velvet contig sequence was used to close the gap between the two contigs. In addition, where a Velvet contig mapped unambiguously to a region within a CA contig, but the alignment indicated a homopolymer run-length discrepancy, the length of the run in the Velvet contig was substituted in the contig. A summary of the final assembly statistics is given in Supplementary Table 2.
Chromosome visualization.
The FISH methodology was performed as previously described48. Details of this method are available from T.M. Davis.
Gene prediction.
We used a new version of the self-training algorithm GeneMark-ES49 to generate a set of ab initio gene predictions for the F. vesca genome. The GeneMark-ES+ software tool was designed for application to genome annotation projects when transcriptome sequence is available. The details of these processes are presented in the Supplementary Note.
Gene homology analysis.
The Inparanoid algorithm50 was used to find orthologous genes and paralogous genes that arise by duplication events. Ortholog clusters were seeded with a two-way best pairwise match, after which an algorithm for adding in-paralogs was applied. First, the all-versus-all (reciprocal) BLAST search was run using sequence files from any two given species pairs (for example, A and B). Sequence pairs with mutual best hits were detected. Sequences from out-group species were used to detect cases of selective loss of orthologs. The A-B sequence pairs were eliminated if either sequence A or sequence B scored higher to an out-group sequence than to each other. Additionally, orthologs based on in-paralogs were clustered together with each remaining pair of potential orthologs. Overlapping clusters were resolved by a set of rules adopted from Inparanoid. The parameters used were the defaults of a cutoff bit score of 50, in-paralog confidence cutoff of 0.05 and group overlap of 0.5, where homologs were grouped if they were similar enough to each other. Other parameters used were a cutoff of 0.5 for total sequence overlaps and 0.25 for segment overlaps. Additional details and specific species analyzed are presented in the Supplementary Note.
Transcriptome analysis.
Total RNA was isolated from mature F. vesca Hawaii-4 (accession PI551572) organs subjected to a wide array of treatment conditions, including stress, light, growth regulators and pharmacological treatments, as previously described51. Normalized pools were converted to full-length enriched cDNA using the SMART method52 and sequenced using Illumina protocols. Reads were filtered and mapped to the F. vesca genome using HashMatch (see URLs) and Supersplat53. Data are available in the NCBI Sequence Read Archive under accession SRA026313.1. Transcript reads used for gene annotation were sequenced using 454, from RNA isolated specifically from fruit (developmental stages from initial buds to overripe) and roots (including root tips, roots and crown root initials). Data are available in the NCBI Sequence Read Archive under accession SRA026350.1. Overrepresented gene ontology categories were determined as described in the Supplementary Note.
Multiple genome alignment.
Genomes from other species were pairwise compared to the F. vesca sequence using MUMmer3, option promer, using the F. vesca genome as the first member of the pair. Results were then merged based on overlaps between matched regions in the anchor genome. Details on genomes used and additional parameters of the test are presented in the Supplementary Note.
Analysis of large duplications in the Fragaria vesca genome.
A comparison of the F. vesca genome was made against itself using MUMmer47 version 3.22 at the nucleotide level (nucmer). Two approaches were implemented. The first measured matches by MUMmer excluding whole contig self-matches. The second used BOWTIE to match Illumina reads against contigs and for quantifying unique hits. Details are presented in the Supplementary Note.
Gene ontology annotation.
A functional annotation pipeline provided gene ontology assignments to 25,051 genes. Peptide sequences were analyzed through Interproscan54, followed by SignalP55, Predotar56 and TMHMM57. Additional details are presented in the Supplementary Note.
Identification of disease resistance genes.
Putative disease resistance genes such as pathogenesis-related (PR) proteins and biosynthesis and/or response genes associated with jasmonic acid, salicylic acid and nitric acid were identified using a keyword search in the NCBI database, with priority given to results from the Rosaceae family. Sequences from this search were used to interrogate the F. vesca genome using BLASTx. Hits with an e value <107, a score > 150 and >50% identity are presented.
Transcription factor identification.
Protein sequences for plant transcription factors were downloaded from the Peking University Plant Transcription Factor Databases repository. A Reciprocal Best Hit (RBH) approach was used to identify and classify putative strawberry transcription factors by BLASTx58 using a threshold cut off of 10 × 10−20. RBHs were extracted by parsing BLASTx tabular outputs using a PERL script. Protein family motifs, detected by InterproScan version 4.4 (ref. 59), were used to confirm (but not exclude) putative strawberry transcription factors. Protein sequence alignments were performed using ClustalW version 2.0.1160. This conservative approach revealed 1,616 transcription factor genes, which is more than Vitis (1,403) but less than Arabidopsis (1,856) (Supplementary Table 17).
Angiosperm phylogeny based on 154 protein-coding genes.
Putative single-copy genes were retrieved from ten angiosperm genomes using the GeneTrees database61 and subsequently available genomes. Homologous protein sequences were trimmed to eliminate non-conserved terminal regions and aligned with MUSCLE62,63, and trees were inferred for each gene family using RAxML 2.3 (ref. 64). Screening for phylogenetic coherence and the presence of paralogs (Supplementary Note) resulted in 154 orthologous gene families missing members from at most two genomes. These families were realigned (MUSCLE) and poorly aligned regions were filtered with Gblocks65. Filtered alignments were concatenated to a matrix of 68,526 characters and analyzed with RAxML using GTR and gamma-distributed rate variation. The approximately unbiased test35, as implemented in the program CONSEL66, was used to evaluate our placement of poplar, the 'poplar-Brassicales topology', against the widely accepted 'Fabidae topology'37.
Sources for the protein or nucleotide datasets, individual sequence identifiers with subsequence ranges, output files from tree analyses and a detailed work flow are available at http://staff.vbi.vt.edu/allan/StrawberryPhylogenomics.
Accession codes.
This whole-genome shotgun project has been deposited at DDBJ, EMBL and GenBank under the project accession AEMH00000000. Sequence reads have been deposited to the short-read archive under the following notation: SRA020125 contains 454-generated genomic reads, SRA026313 contains Illumina RNA-seq and genomic data and SRA026350 contains 454 transcriptome reads.
Accession codes
Accessions
NCBI Reference Sequence
Sequence Read Archive
References
Darrow, G.M. The Strawberry: History, Breeding and Physiology. (Holt, Rinehart and Winston, New York, New York, USA, 1966).
Shulaev, V. et al. Multiple models for Rosaceae genomics. Plant Physiol. 147, 985–1003 (2008).
Alsheikh, M.K., Suso, H.P., Robson, M., Battey, N.H. & Wetten, A. Appropriate choice of antibiotic and Agrobacterium strain improves transformation of antibiotic-sensitive Fragaria vesca and F.v. semperflorens. Plant Cell Rep. 20, 1173–1180 (2002).
Oosumi, T. et al. High-efficiency transformation of the diploid strawberry (Fragaria vesca) for functional genomics. Planta 223, 1219–1230 (2006).
Oosumi, T., Ruiz-Rojas, J.J., Veilleux, R.E., Dickerman, A. & Shulaev, V. Implementing reverse genetics in Rosaceae: analysis of T-DNA flanking sequences of insertional mutant lines in the diploid strawberry, Fragaria vesca L. Physiol. Plant. 140, 1–9 (2010).
Ruiz-Rojas, J.J. et al. SNP discovery and genetic mapping of T-DNA insertional mutants in Fragaria vesca L. Theor. Appl. Genet. 121, 449–463 (2010).
Sargent, D.J. et al. The development of a bin mapping population and the selective mapping of 103 markers in the diploid Fragaria reference map. Genome 51, 120–127 (2008).
Davis, T.M. & Yu, H. A linkage map of the diploid strawberry, Fragaria vesca. J. Hered. 88, 215–221 (1997).
Lim, K.Y. Karyotype and ribosomal gene mapping in Fragaria vesca L. Acta Hortic. 649, 103–106 (2004).
Cabrera, A. et al. Development and bin mapping of a Rosaceae Conserved Ortholog Set (COS) of markers. BMC Genomics 10, 562 (2009).
Potter, D. et al. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266, 5–43 (2007).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Rice Chromosomes 11 and 12 Sequencing Consortia. The sequence of rice chromosomes 11 and 12, rich in disease resistance genes and recent gene duplications. BMC Biol. 3, 20 (2005).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Velasco, R. et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 42, 833–839 (2010).
Schnable, P.S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009).
Baucom, R.S. et al. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet. 5, e1000732 (2009).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Mewes, H.W. et al. MIPS: analysis and annotation of proteins from whole genomes. Nucleic Acids Res. 32, D41–D44 (2004).
Ouyang, S. & Buell, C.R. The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res. 32, D360–D363 (2004).
Davis, T.M. et al. An examination of targeted gene neighborhoods in strawberry. BMC Plant Biol. 10, 81 (2010).
Pontaroli, A.C. et al. Gene content and distribution in the nuclear genome of Fragaria vesca. Plant Genome 2, 93–101 (2009).
Folta, K.M. et al. A.transcript accounting from diverse tissues of a cultivated strawberry. Plant Genome 3, 90–105 (2010).
Aharoni, A. & O′Connell, A.P. Gene expression analysis of strawberry achene and receptacle maturation using DNA microarrays. J. Exp. Bot. 53, 2073–2087 (2002).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y.O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Wachter, A. et al. Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell 19, 3437–3450 (2007).
Daniell, H. et al. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 116, 723–737 (2008).
Paterson, A.H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009).
Klessig, D.F. et al. Nitric oxide and salicylic acid signaling in plant defense. Proc. Natl. Acad. Sci. USA 97, 8849–8855 (2000).
Dempsey, D.A., Silva, H. & Klessig, D.F. Engineering disease and pest resistance in plants. Trends Microbiol. 6, 54–61 (1998).
Wei, Y.L. et al. Molecular cloning of Brassica napus TRANSPARENT TESTA 2 gene family encoding potential MYB regulatory proteins of proanthocyanidin biosynthesis. Mol. Biol. Rep. 34, 105–120 (2007).
Yoshida, K. et al. Functional differentiation of Lotus japonicus TT2s, R2R3-MYB transcription factors comprising a multigene family. Plant Cell Physiol. 49, 157–169 (2008).
Tian, Y. & Dickerman, A.W. GeneTrees: a phylogenomics resource for prokaryotes. Nucleic Acids Res. 35, D328–D331 (2007).
Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508 (2002).
Williams, K.P. et al. Phylogeny of Gammaproteobacteria. J. Bacteriol. 192, 2305–2314 (2010).
Wang, H. et al. Rosid radiation and the rapid rise of Angiosperm-dominated forests. Proc. Natl. Acad. Sci. USA 106, 3853–3858 (2009).
Zhu, X.Y. et al. Mitochondrial matR sequences help to resolve deep phylogenetic relationships in rosids. BMC Evol. Biol. 7, 217 (2007).
Duarte, J.M. et al. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 10, 61 (2010).
Wurdack, K.J. & Davis, C.C. Malpighiales phylogenetics: gaining ground on one of the most recalcitrant clades in the Angiosperm tree of life. Am. J. Bot. 96, 1551–1570 (2009).
Qui, Y.-L. et al. Angiosperm phylogeny inferred from sequences of four mitochondrial genes. J. Syst. Evol. 48, 391–425 (2010).
Endress, P.K. & Matthews, M.L. First steps towards a floral structural characterization of the major rosid subclades. Plant Syst. Evol. 260, 223–251 (2006).
Renoult, J.P., Kjellberg, F., Grout, C., Santoni, S. & Khadari, B. Cyto-nuclear discordance in the phylogeny of Ficus section Galoglychia and host shifts in plant-pollinator associations. BMC Evol. Biol. 9, 248 (2009).
Rokas, A., Williams, B.L., King, N. & Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425, 798–804 (2003).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Zerbino, D.R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Liu, B. et al. Molecular cytogenetic analysis of four Larix species by bicolor fluorescence in situ hybridization and dapi banding. Int. J. Plant Sci. 167, 367–372 (2006).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y.O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Ostlund, G. et al. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 38, D196–D203 (2010).
Folta, K.M. et al. A.transcript accounting from diverse tissues of a cultivated strawberry. Plant Genome 3, 90–105 (2010).
Zhu, Y.Y., Machleder, E.M., Chenchik, A., Li, R. & Siebert, P.D. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 30, 892–897 (2001).
Bryant, D.W. Jr., Shen, R., Priest, H.D., Wong, W.K. & Mockler, T.C. Supersplat-spliced RNA-seq alignment. Bioinformatics 26, 1500–1505 (2010).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Emanuelsson, O., Brunak, S., von Heijne, G. & Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971 (2007).
Small, I., Peeters, N., Legeai, F. & Lurin, C. Predotar: A tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4, 1581–1590 (2004).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Zdobnov, E.M. & Apweiler, R. InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Larkin, M.A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Tian, Y. & Dickerman, A.W. GeneTrees: a phylogenomics resource for prokaryotes. Nucleic Acids Res. 35, D328–D331 (2007).
Edgar, R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 1–19 (2004).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A., Ludwig, T. & Meier, H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463 (2005).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Shimodaira, H. & Hasegawa, M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247 (2001).
Acknowledgements
This work was supported by Roche and 454 Sequencing; the Virginia Bioinformatics Institute; the University of Florida Institute of Food and Agricultural Sciences (IFAS) Dean for Research; the University of Florida Strawberry Breeding Program; The Province of Trento, Italy (to R.V.); Driscoll's Strawberry Associates; United States Department of Agriculture/Cooperative State Research, Education and Extension Service (USDA/CSREES) Hatch Project VA-135816 (to B.F.); Rutgers Busch Biomedical Funding (to T.P.M.); East Malling Trust (EMT) and Biotechnology and Biological Sciences Research Council (BBSRC) (to D.J.S. and E.L.G.); the Oregon State Agricultural Research Foundation #ARF4435 (to T.C.M.); the Oregon State Computational and Genome Biology Initiative (to T.C.M.); Oregon State University start-up fund (to P.J.); the Center for Genomics and Bioinformatics, supported in part by the METACyt Initiative of Indiana University (to K.M.); US National Institutes of Health (grant HG00783; to M.B.); USDA-CSREES National Research Initiative (NRI) Plant Genome Grant 2008-35300-04411 and New Hampshire Agricultural Experiment Station Project NH00535 (to T.M.D.); and USDA/ARS CRIS #1275-21000-180-01R (to J.P.S.).
Author information
Authors and Affiliations
Contributions
Project management: K.M.F., V.S., R.E.V.
Project coordination: O.F., T.C.M., D.J.S., T.M.D., J.P.S., N.B., T.-L.A., L.M., H.S., A.C.A., R.N.C., T.P.M.
Germplasm, DNA and RNA preparation: T.P.M., J.P.S., A.Z., D.Y.S., K.M.F., S.H.H.
Library construction and sequencing: O.F., T.C.M., R.V.J., C.E., T.H., J.C., K.M., C.K., B.D., O.R.C., M.T., R. Velasco, J.D., S.A.F., T.P.M., S.E.F., R.P.H., B.F., R.A.K.,W.W.
Sequence processing and assembly: A.L.D., S.L.S., M.T., S.P.M., R. Velasco, R. Viola, T.C.M., H.D.P., D.W.B., R.P.H., A.L., S.F., T.P.M.
Anchoring scaffolds to linkage map: D.J.S., J.-M.C., J.G.R., A.C., J.J.R.R., E.L.G., M.T., R. Velasco, T.M.D., B.L., T.-L.A., B.D.-R., A.M., P.A.
Computational resources (GBrowse, Blast, Genbank submission and data management): R.N.C., S.P.M., S.A.G., H.D.P., L.J.W.
Gene prediction and annotation: P.B., M.B., T.C.M., H.D.P., D.W.B., R.N.C., R.P.H., N.B., J.P.S., S.F., A.C.A., K.P.W.
Gene ontology and pathway analysis: P.J., T.C.M., P.D., J.E., R.R., S.N.
Evolutionary analyses: A.L., A.W.D., D.J.S.
Comparative genomics: D.J.S., A.L., J.C.S., E.L.G., M.C., K.M.F.
Analysis of gene families: A.C.A., A. Adato, A. Aharoni.
Contributed tables, figures and other analyses: H.S., L.M., T.C.M., D.J.S., B.L., T.M.D., W.S., A.L., P.J., H.W., J.L.B., R.E.V.
Provided funding and/or other support: V.S., R.E.V., R. Velasco, R. Viola, K.M.F., T.C.M., C.E., J.G.R., J.P.S., K.M., S.S.K., R.P.H., B.F., R.M.
Manuscript preparation: K.M.F., R.E.V., T.M.D., T.-L.A., J.P.S., A.L., N.B., D.J.S., T.C.M., P.J., A.C.A., V.S., K.M., J.C.S., H.S., L.M., A. Adato, H.W., S.S.K., A. Aharoni, J.L.B., R. Velasco.
Contributed to revisions: T.C.M., R.E.V., K.M., T.M.D., J.P.S., M.B., N.B., T.-L.A., H.S., L.M., K.M.F.
All authors critically read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Tables 1–18 and Supplementary Figures 1–10 (PDF 3059 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar license.
About this article
Cite this article
Shulaev, V., Sargent, D., Crowhurst, R. et al. The genome of woodland strawberry (Fragaria vesca). Nat Genet 43, 109–116 (2011). https://doi.org/10.1038/ng.740
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.740
This article is cited by
-
Chromosome-scale genome assembly and annotation of Cotoneaster glaucophyllus
Scientific Data (2024)
-
Effect of different growing media on pomological and phytochemical parameters of Fragaria vesca ʻYellow Wonderʼ and Fragaria ×ananassa ʻCamarosaʼ: a comparative study
BMC Plant Biology (2023)
-
Genome-wide identification and expression analysis of the coronatine-insensitive 1 (COI1) gene family in response to biotic and abiotic stresses in Saccharum
BMC Genomics (2022)
-
Computational analysis of potential candidate genes involved in the cold stress response of ten Rosaceae members
BMC Genomics (2022)
-
Exploring machine learning: a scientometrics approach using bibliometrix and VOSviewer
SN Applied Sciences (2022)
