
Abstract
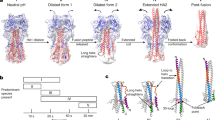
Alphaviruses are enveloped RNA viruses that have a diameter of about 700 Å and can be lethal human pathogens1. Entry of virus into host cells by endocytosis is controlled by two envelope glycoproteins, E1 and E2. The E2–E1 heterodimers form 80 trimeric spikes on the icosahedral virus surface1,2, 60 with quasi-three-fold symmetry and 20 coincident with the icosahedral three-fold axes arranged with T = 4 quasi-symmetry. The E1 glycoprotein has a hydrophobic fusion loop at one end and is responsible for membrane fusion3,4. The E2 protein is responsible for receptor binding5,6 and protects the fusion loop at neutral pH. The lower pH in the endosome induces the virions to undergo an irreversible conformational change in which E2 and E1 dissociate and E1 forms homotrimers, triggering fusion of the viral membrane with the endosomal membrane and then releasing the viral genome into the cytoplasm3,4. Here we report the structure of an alphavirus spike, crystallized at low pH, representing an intermediate in the fusion process and clarifying the maturation process. The trimer of E2–E1 in the crystal structure is similar to the spikes in the neutral pH virus except that the E2 middle region is disordered, exposing the fusion loop. The amino- and carboxy-terminal domains of E2 each form immunoglobulin-like folds, consistent with the receptor attachment properties of E2.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Accession codes
Primary accessions
Protein Data Bank
Data deposits
The atomic coordinates of the E2–E1 heterodimer crystal structures have been deposited with the Protein Data Bank (accession number 3MUU). The fit of the E2–E1 heterodimer into the cryo-EM reconstruction of Sindbis virus has been deposited with the Protein Data Bank (accession number 3MUW).
References
Strauss, J. H. & Strauss, E. G. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58, 491–562 (1994)
von Bonsdorff, C. H. & Harrison, S. C. Sindbis virus glycoproteins form a regular icosahedral surface lattice. J. Virol. 16, 141–145 (1975)
Kielian, M. Membrane fusion and the alphavirus life cycle. Adv. Virus Res. 45, 113–151 (1995)
White, J. & Helenius, A. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc. Natl Acad. Sci. USA 77, 3273–3277 (1980)
Dubuisson, J. & Rice, C. M. Sindbis virus attachment: isolation and characterization of mutants with impaired binding to vertebrate cells. J. Virol. 67, 3363–3374 (1993)
Strauss, E. G., Stec, D. S., Schmaljohn, A. L. & Strauss, J. H. Identification of antigenically important domains in the glycoproteins of Sindbis virus by analysis of antibody escape variants. J. Virol. 65, 4654–4664 (1991)
Lescar, J. et al. The fusion glycoprotein shell of Semliki Forest Virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105, 137–148 (2001)
Mukhopadhyay, S. et al. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure 14, 63–73 (2006)
Zhang, W. et al. Placement of the structural proteins in Sindbis virus. J. Virol. 76, 11645–11658 (2002)
Schmidt, T. G. & Skerra, A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nature Protocols 2, 1528–1535 (2007)
Smit, J. M., Bittman, R. & Wilschut, J. Low-pH-dependent fusion of Sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J. Virol. 73, 8476–8484 (1999)
White, J., Kartenbeck, J. & Helenius, A. Fusion of Semliki Forest virus with the plasma membrane can be induced by low pH. J. Cell Biol. 87, 264–272 (1980)
Voss, J. E. et al. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 10.1038/nature09555 (this issue)
Meyer, W. J. & Johnston, R. E. Structural rearrangement of infecting Sindbis virions at the cell surface: mapping of newly accessible epitopes. J. Virol. 67, 5117–5125 (1993)
Zhang, W., Heil, M., Kuhn, R. J. & Baker, T. S. Heparin binding sites on Ross River virus revealed by electron cryo-microscopy. Virology 332, 511–518 (2005)
Wu, S.-R. et al. The dynamic envelope of a fusion class II virus. Prefusion stages of Semliki Forest virus revealed by electron cryomicroscopy. J. Biol. Chem. 282, 6752–6762 (2007)
Griffin, D. E. Roles and reactivities of antibodies to alphaviruses. Semin. Virol. 6, 249–255 (1995)
Wahlberg, J. M., Boere, W. A. & Garoff, H. The heterodimeric association between the membrane proteins of Semliki Forest virus changes its sensitivity to low pH during virus maturation. J. Virol. 63, 4991–4997 (1989)
Ziemiecki, A. & Garoff, H. Subunit composition of the membrane glycoprotein complex of Semliki Forest virus. J. Mol. Biol. 122, 259–269 (1978)
Ziemiecki, A., Garoff, H. & Simons, K. Formation of the Semliki Forest virus membrane glycoprotein complexes in the infected cell. J. Gen. Virol. 50, 111–123 (1980)
Ferlenghi, I. et al. The first step: activation of the Semliki Forest virus spike protein precursor causes a localized conformational change in the trimeric spike. J. Mol. Biol. 283, 71–81 (1998)
Paredes, A. M. et al. Structural localization of the E3 glycoprotein in attenuated Sindbis virus mutants. J. Virol. 72, 1534–1541 (1998)
Wu, S. R., Haag, L., Sjoberg, M., Garoff, H. & Hammar, L. The dynamic envelope of a fusion class II virus. E3 domain of glycoprotein E2 precursor in Semliki Forest virus provides a unique contact with the fusion protein E1. J. Biol. Chem. 283, 26452–26460 (2008)
Zhang, X. & Kielian, M. Mutations that promote furin-independent growth of Semliki Forest virus affect p62–E1 interactions and membrane fusion. Virology 327, 287–296 (2004)
Heidner, H. W., McKnight, K. L., Davis, N. L. & Johnston, R. E. Lethality of PE2 incorporation into Sindbis virus can be suppressed by second-site mutations in E3 and E2. J. Virol. 68, 2683–2692 (1994)
Wahlberg, J. M. & Garoff, H. Membrane fusion process of Semliki Forest virus. I. Low pH-induced rearrangement in spike protein quaternary structure precedes virus penetration into cells. J. Cell Biol. 116, 339–348 (1992)
Gibbons, D. L. et al. Conformational change and protein–protein interactions of the fusion protein of Semliki Forest virus. Nature 427, 320–325 (2004)
Li, L. et al. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319, 1830–1834 (2008)
Yu, I. M. et al. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319, 1834–1837 (2008)
Rossmann, M. G., Bernal, R. & Pletnev, S. V. Combining electron microscopic with X-ray crystallographic structures. J. Struct. Biol. 136, 190–200 (2001)
Owen, K. E. & Kuhn, R. J. Identification of a region in the Sindbis virus nucleocapsid protein that is involved in specificity of RNA encapsidation. J. Virol. 70, 2757–2763 (1996)
Lehr, R. V., Elefante, L. C., Kikly, K. K., O'Brien, S. P. & Kirkpatrick, R. B. A modified metal-ion affinity chromatography procedure for the purification of histidine-tagged recombinant proteins expressed in Drosophila S2 cells. Protein Expr. Purif. 19, 362–368 (2000)
Sharff, A. J., Koronakis, E., Luisi, B. & Koronakis, V. Oxidation of selenomethionine: some MADness in the method!. Acta Crystallogr. D 56, 785–788 (2000)
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997)
Collaborative Computational Project Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 (1994)
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007)
Sheldrick, G. M. A short history of SHELX. Acta Crystallogr. A 64, 112–122 (2008)
de La Fortelle, E. & Bricogne, G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 276, 472–494 (1997)
Cowtan, K. D. ‘dm’: an automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newsl. Protein Crystallogr. 31, 34–38 (1994)
Terwilliger, T. C. Maximum-likelihood density modification. Acta Crystallogr. D 56, 965–972 (2000)
Kleywegt, G. J. & Jones, T. A. Template convolution to enhance or detect structural features in macromolecular electron-density maps. Acta Crystallogr. D 53, 179–185 (1997)
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 (1997)
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Winn, M. D., Isupov, M. N. & Murshudov, G. N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D 57, 122–133 (2001)
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Acknowledgements
We wish to thank S. Sun, A. Aksyuk and T. Edwards for discussions. We are also grateful to S. Kelly for help in the preparation of the manuscript. We thank F. Rey for sharing the coordinates of Chikungunya virus E2–E1 to help interpret the Sindbis virus cryo-EM density of the E2 domain B. We would like to thank the staff at the Advanced Photon Source, Argonne National Laboratory, GM/CA sector for their help in data collection. The work was supported by NIH grant P01 AI055672 to R.J.K. and M.G.R.
Author information
Authors and Affiliations
Contributions
L.L. designed the expression constructs, J.J. cloned the constructs, and L.L. and J.J. developed the expression system and purified the protein. L.L. crystallized the protein, collected X-ray diffraction data and, with Y.X., determined the structure. L.L., R.J.K. and M.G.R. discussed the results and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
The file contains Supplementary Tables 1-4, additional references and Supplementary Figures 1-7 with legends. (PDF 598 kb)
Rights and permissions
About this article
Cite this article
Li, L., Jose, J., Xiang, Y. et al. Structural changes of envelope proteins during alphavirus fusion. Nature 468, 705–708 (2010). https://doi.org/10.1038/nature09546
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature09546
This article is cited by
-
LDLR is used as a cell entry receptor by multiple alphaviruses
Nature Communications (2024)
-
STT3A-mediated viral N-glycosylation underlies the tumor selectivity of oncolytic virus M1
Oncogene (2023)
-
A molecular understanding of alphavirus entry and antibody protection
Nature Reviews Microbiology (2023)
-
Antigenicity and immunogenicity of chikungunya virus-like particles from mosquito cells
Applied Microbiology and Biotechnology (2023)
-
The evolution of chikungunya virus circulating in Indonesia: Sequence analysis of the orf2 gene encoding the viral structural proteins
International Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
