
Abstract
Lynch syndrome is the most common inherited colorectal cancer syndrome, accounting for 2–4% of all colorectal cancer cases. This review focuses on the tissue workup of Lynch syndrome, including methods to determine whether or not a tumor is mismatch repair deficient, and whether a mismatch repair-deficient tumor is sporadic or Lynch syndrome-associated. Strategies for determining which tumors to test and how best to implement a Lynch syndrome screening program are also discussed, as well as potential developments in the future.
Similar content being viewed by others

Main
Lynch syndrome is a dominantly inherited cancer predisposition syndrome characterized by an increased risk of numerous cancers, including colorectal, endometrial, ovarian, renal pelvis, ureter, small intestine, stomach, hepatobiliary tract, and pancreas. The markedly increased risk of colorectal cancer, usually proximally located, accounts for its original name, hereditary non-polyposis colorectal cancer, or HNPCC. The renaming of the syndrome to Lynch is in part due to the recognition of the risk of these extra-colonic cancers; indeed, among women (and in certain affected families) the risk of endometrial cancer may be greater than the risk of colorectal cancer.1
Mismatch repair deficiency
Lynch syndrome-associated tumors are mismatch repair deficient. Mistakes commonly occur on the newly synthesized strand of DNA during DNA replication. Complexes of mismatch repair proteins recognize the resultant mismatches between the newly synthesized strand and the original DNA strand and correct them. In mismatch repair-deficient tumors these mismatches are not corrected, leading to the accumulation of mutations and, ultimately, neoplasia. However, a minority of sporadic tumors not associated with Lynch syndrome also exhibit mismatch repair deficiency, including 10–15% of colorectal cancers. An important part of the workup of Lynch syndrome involves differentiating sporadic from Lynch syndrome-associated mismatch repair-deficient tumors.
The two main clinical tests for the detection of mismatch repair deficiency are microsatellite instability by PCR and immunohistochemistry (IHC) for mismatch repair proteins. Microsatellite repeats are short-nucleotide repeats that are especially prone to inaccurate DNA replication. In mismatch repair-deficient tumors these replication mistakes are not corrected, leading to expansion or contraction of these repeats, or microsatellite instability. Most clinical tests of microsatellite instability use a panel of mononucleotide repeats, as these are probably the most sensitive and specific markers for mismatch repair deficiency.2, 3, 4 Most microsatellite repeats are in noncoding regions of DNA, although there are exceptions.
IHC for the mismatch repair proteins MLH1, MSH2, MSH6, and PMS2 is also a fairly sensitive and specific indicator of mismatch repair deficiency. Mismatch repair-proficient tumors will show nuclear staining for all four proteins. Mismatch repair-deficient tumors will show loss (typically complete) of nuclear staining for one or more of the proteins. The IHC profile helps determine which gene may be responsible for the mismatch repair deficiency. If nuclear staining of MLH1 and PMS2 is lost, then MLH1 is likely defective. This is because the stability of PMS2 depends upon its ability to form a complex with MLH1; loss of MLH1 therefore results in secondary loss of PMS2. Similarly, if MSH2 and MSH6 are lost, then MSH2 is likely defective, as MSH6 is unstable without MSH2. The stability of MLH1 and MSH2 are not as dependent on these complexes, and so isolated loss of MSH6 or PMS2 usually indicates a defect in MSH6 or PMS2, respectively.5, 6 It should be noted that there are exceptions to these IHC profile/gene associations. For example, germline MLH1 mutations may be associated with isolated loss of PMS2 by IHC.7
MSI vs IHC
Microsatellite instability by PCR and IHC for mismatch repair proteins should be viewed as complementary tests; while both are sensitive and specific for mismatch repair deficiency, neither is perfect, and both will miss some mismatch repair-deficient tumors.2, 5 One major advantage of IHC over PCR is that IHC provides information regarding the relevant gene(s); this is important in subsequent tests in the workup of Lynch syndrome.
Like all IHC tests, there may be problems in interpretation of IHC for mismatch repair proteins. Staining variability within a tumor may be an issue, with some areas of the tumor that stain and others that do not. In mismatch repair-proficient tumors this staining typically parallels the internal control staining of non-neoplastic cells, indicating a fixation or processing issue rather than an actual protein loss. Our practice is to only score IHC loss in a tumor in areas of the slide in which immunostaining of internal control non-neoplastic nuclei is maintained. If tumor staining is lost, but there is no internal control staining, we state that the IHC result is uninterpretable.
Another IHC problem is decreased MSH6 staining after chemoradiation, a particular problem with rectal cancers.8 Staining of the pretreatment biopsy may be helpful in such cases to determine whether the MSH6 loss is real. ‘Clonal’ or focal MSH6 loss may also occur (unrelated to chemoradiation), in which some areas of the tumor show intact MSH6 staining and other areas show loss (with intact internal control staining). This is due to the fact that there is a coding mononucleotide repeat in MSH6 that may become unstable and inactivate MSH6 in a mismatch repair-deficient tumor; those areas of the tumor in which the MSH6 mononucleotide repeat is unstable will lose immunostaining, while those areas in which the mononucleotide repeat does not become unstable will show intact staining.9 Importantly, this ‘clonal’ MSH6 loss typically occurs in tumors that are mismatch repair deficient due to causes unrelated to MSH6, including sporadic mismatch repair-deficient tumors or tumors with germline mutations of mismatch repair genes besides MSH6.
Finally, there are occasional cases in which IHC staining for one or more proteins is markedly reduced (with maintained internal control staining), but not completely lost. In such instances we recommend reporting the markedly decreased staining, stating that this suggests the possibility of mismatch repair deficiency, and recommend testing the tumor for microsatellite instability by PCR.
Mismatch repair-deficient tumor: sporadic or Lynch syndrome?
Once a tumor has been shown to be mismatch repair deficient, either by IHC or by PCR, the next step is to determine whether the tumor may be associated with Lynch syndrome or is sporadic. The molecular basis for sporadic mismatch repair-deficient tumors is usually acquired methylation of the MLH1 promoter; this turns off the production of MLH1 mRNA and, therefore, the MLH1 protein.6 The IHC profile associated with a sporadic mismatch repair-deficient tumor is therefore usually the loss of MLH1 and PMS2. However, a germline mutation of MLH1 in Lynch syndrome will also be associated with this IHC profile. The other IHC profiles, combined MSH2/MSH6 loss or isolated loss of MSH6 or PMS2, are more likely to be associated with Lynch syndrome due to a germline mutation in one of these genes, and germline mutational analysis (usually of leukocyte DNA from blood) is indicated. It should be noted that occasional cases of MSH2/MSH6 loss are due to deletions of the 3′ end of EPCAM, a gene just upstream of MSH2.10 However, this is well known to geneticists, and the germline evaluation of MSH2 typically includes a test for EPCAM deletions.
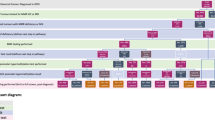
Sporadic mismatch repair-deficient tumors are actually more common than Lynch syndrome-associated mismatch repair-deficient tumors; the most common IHC result in a mismatch repair-deficient tumor is combined loss of MLH1 and PMS2. If tumors associated with this immunohistochemical profile can be shown to be sporadic, then costly germline evaluation of MLH1 can be avoided and the patient and family members do not have to be followed up for Lynch-associated tumors. Fortunately, there are molecular/immunohistochemical features of the tumor that can be used to suggest whether an IHC profile of combined MLH1/PMS2 loss is associated with a sporadic mismatch repair-deficient tumor or with Lynch syndrome. MLH1 methylation is the basis for most sporadic mismatch repair-deficient tumors, and it is only rarely seen in Lynch syndrome. The BRAF V600E mutation is seen in ∼50% of sporadic mismatch repair-deficient colorectal tumors; this mutation is almost never seen in Lynch-associated tumors.6 Unfortunately, mutations in BRAF are uncommon in extra-colonic sporadic tumors,11 and therefore BRAF testing is not useful in distinguishing sporadic mismatch repair-deficient extra-colonic tumors from those associated with Lynch syndrome. A reasonable approach to a colorectal tumor with an IHC profile of combined MLH1/PMS2 loss is to test for the BRAF V600E mutation; if this is present, then the tumor is likely to be sporadic and germline testing of MLH1 can usually be avoided. An antibody specific for the BRAF V600E mutation has been developed, potentially making the tissue evaluation of Lynch syndrome for the most part an IHC process.12 However, we still consider molecular methods to be the more objective and definitive evaluation for the BRAF mutation, as these avoid the staining variability and the somewhat subjective interpretation inherent with most IHC stains. If the colorectal cancer does not show the BRAF mutation, or the tumor is extra-colonic, then MLH1 promoter methylation may be evaluated. MLH1 promoter methylation indicates that the tumor is likely to be sporadic and that MLH1 germline testing can be avoided. As mentioned above, there are rare Lynch syndrome tumors with BRAF mutations or MLH1 promoter methylation, so the presence of either of these alterations does not completely exclude the possibility of Lynch syndrome. This algorithmic approach to tissue testing for colorectal cancer is shown in Figure 1.

Algorithmic approach to the tissue workup of Lynch syndrome starting with immunohistochemistry (IHC) for mismatch repair (MMR) proteins. Note that the BRAF V600E mutation test is not applicable to extra-colonic cancers, such as endometrial tumors.
Which tumors should be tested?
There are several approaches to determine which tumors to test for mismatch repair deficiency. One is the Revised Bethesda Guidelines, which takes into account numerous factors, including the age of the individual, the histology of the tumor, and a personal and family history of Lynch syndrome-associated tumors.13 It should be noted that much of the information regarding family history is not available to the pathologist, and it is therefore very difficult to follow these guidelines without close collaboration with a clinician, such as a genetic counselor. It has also been estimated that even if properly applied these guidelines will miss nearly 30% of Lynch-associated tumors.14
Another approach is universal screening, in which every colorectal (and endometrial) cancer is tested for mismatch repair deficiency, typically by IHC. The rationale behind this approach is that individuals may present with Lynch syndrome at older ages,15 the family history of Lynch-associated tumors is not always available, and that the histological features of mismatch repair deficiency, while fairly robust,16 are not perfect, and IHC is a good and fairly cheap screening test. A third approach is the Jerusalem criteria, in which all colorectal cancers in individuals under the age of 70 are tested. This has been estimated to miss a little over 10% of Lynch syndrome.17
Adenomatous polyps are the precursor for Lynch syndrome-associated colorectal cancers, and mismatch repair deficiency has been detected in adenomas from individuals with Lynch syndrome. However, not all Lynch syndrome-associated adenomas will exhibit mismatch repair deficiency; indeed, a recent study reported that half of these adenomas are not mismatch repair deficient, and that polyps larger than 8–10 mm were much more likely to exhibit mismatch repair deficiency.18 Another study found that 80% of Lynch-associated adenomas were mismatch repair deficient by IHC.19 This study identified a relationship between mismatch repair deficiency and a villous component or high-grade dysplasia, but not with size. At this point the available evidence suggests that mismatch repair deficiency in an adenoma is useful in identifying potential Lynch syndrome mutation carriers, but that the lack of mismatch repair deficiency in an adenoma is not as strong a criterion to exclude Lynch syndrome as the lack of mismatch repair deficiency in a cancer.
Utilizing a team approach
A ‘team’ approach is very important in the clinical workup of Lynch syndrome. The choice of a screening method to test tumors and the actual tissue testing are important, but they will not be clinically useful if these results are not adequately communicated to a clinician who understands and can act upon the results. In our experience collaboration with genetic counselors has been extremely useful, and it has been shown that reporting results to genetic counselors results in a higher likelihood of appropriate follow-up and genetic testing.20 Also, these clinicians can suggest additional testing outside the usual algorithm if the index of suspicion for Lynch syndrome is high. This may involve testing microsatellite instability by PCR even if the IHC is normal, as we know that neither IHC nor PCR detects all mismatch repair-deficient tumors. It may also involve sequencing genes even if BRAF is mutated or the MLH1 promoter is methylated, since Lynch syndrome-associated tumors may rarely exhibit these alterations.
The future
As the cost of next-generation sequencing (also known as massively parallel sequencing) continues to decrease, it is certainly possible that germline sequencing of panels of cancer-related genes (including all four mismatch repair genes) will be more cost-effective than the tissue workup described in this review. However, there may still be a role for some of these tissue evaluations, especially IHC for mismatch repair proteins. One reason for this is that IHC alone is a very cheap test and will show most colon cancers to be mismatch repair proficient, effectively excluding these individuals from further consideration for Lynch syndrome. Another reason is the fact that sequencing of numerous genes predictably leads to the detection of variants of unknown significance, genetic changes (such as amino-acid substitutions) in which it is difficult to determine whether or not the alteration is actually deleterious to the protein and may therefore be associated with the disease. In such cases the IHC profile may contribute to the evaluation of the significance of the variant. For example, a variant of unknown significance in MSH6 is probably not pathogenic if the IHC profile is loss of MLH1 and PMS2. This application, however, may become less useful as pathogenicity prediction models improve and databases of pathogenic and non-pathogenic alterations continue to grow.
References
Jasperson KW, Tuohy TM, Neklason DW et al. Hereditary and familial colon cancer. Gastroenterology 2010;138:2044–2058.
Zhang L . Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: part II. The utility of microsatellite instability testing. J Mol Diagn 2008;10:301–307.
You JF, Buhard O, Ligtenberg MJ et al. Tumours with loss of MSH6 expression are MSI-H when screened with a pentaplex of five mononucleotide repeats. Br J Cancer 2010;103:1840–1845.
Cicek MS, Lindor NM, Gallinger S et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J Mol Diagn 2011;13:271–281.
Shia J . Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: part I. The utility of immunohistochemistry. J Mol Diagn 2008;10:293–300.
Geiersbach KB, Samowitz WS . Microsatellite instability and colorectal cancer. Arch Pathol Lab Med 2011;135:1269–1277.
Halvarsson B, Lindblom A, Rambech E et al. The added value of PMS2 immunostaining in the diagnosis of hereditary nonpolyposis colorectal cancer. Fam Cancer 2006;5:353–358.
Bao F, Panarelli NC, Rennert H et al. Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am J Surg Pathol 2010;34:1798–1804.
Shia J, Zhang L, Shike M et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1/PMS2 deficiency. Mod Pathol 2013;26:131–138.
Tutlewska K, Lubinski J, Kurzawski G . Germline deletions in the EPCAM gene as a cause of Lynch syndrome - literature review. Hered Cancer Clin Pract 2013;11:9.
Kawaguchi M, Yanokura M, Banno K et al. Analysis of a correlation between the BRAF V600E mutation and abnormal DNA mismatch repair in patients with sporadic endometrial cancer. Int J Oncol 2009;34:1541–1547.
Toon CW, Walsh MD, Chou A et al. BRAFV600E immunohistochemistry facilitates universal screening of colorectal cancers for Lynch syndrome. Am J Surg Pathol 2013;37:1592–1602.
Umar A, Boland CR, Terdiman JP et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261–268.
Hampel H, Frankel WL, Martin E et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26:5783–5788.
Hampel H, Frankel WL, Martin E et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851–1860.
Greenson JK, Huang SC, Herron C et al. Pathologic predictors of microsatellite instability in colorectal cancer. Am J Surg Pathol 2009;33:126–133.
Boland CR, Shike M . Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology 2010;138:2197 e1–2197 e7.
Yurgelun MB, Goel A, Hornick JL et al. Microsatellite instability and DNA mismatch repair protein deficiency in Lynch syndrome colorectal polyps. Cancer Prev Res (Phila) 2012;5:574–582.
Walsh MD, Buchanan DD, Pearson SA et al. Immunohistochemical testing of conventional adenomas for loss of expression of mismatch repair proteins in Lynch syndrome mutation carriers: a case series from the Australasian site of the colon cancer family registry. Mod Pathol 2012;25:722–730.
Heald B, Plesec T, Liu X et al. Implementation of universal microsatellite instability and immunohistochemistry screening for diagnosing lynch syndrome in a large academic medical center. J Clin Oncol 2013;31:1336–1340.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author may receive royalties in the future related to sale of the Ventana BRAF V600E antibody, and has performed consulting work for Qiagen.
Rights and permissions
About this article

Cite this article
Samowitz, W. Evaluation of colorectal cancers for Lynch syndrome: practical molecular diagnostics for surgical pathologists. Mod Pathol 28 (Suppl 1), S109–S113 (2015). https://doi.org/10.1038/modpathol.2014.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.127
This article is cited by
-
CMTM6 expression in M2 macrophages is a potential predictor of PD-1/PD-L1 inhibitor response in colorectal cancer
Cancer Immunology, Immunotherapy (2021)
-
Discordant DNA mismatch repair protein status between synchronous or metachronous gastrointestinal carcinomas: frequency, patterns, and molecular etiologies
Familial Cancer (2021)
-
Prognostic perspectives of PD-L1 combined with tumor-infiltrating lymphocytes, Epstein-Barr virus, and microsatellite instability in gastric carcinomas
Diagnostic Pathology (2020)
-
Correlations between microsatellite instability and the biological behaviour of tumours
Journal of Cancer Research and Clinical Oncology (2019)
-
Current applications of molecular pathology in colorectal carcinoma
Applied Cancer Research (2017)
