
Abstract
Epithelioid morphology, mimicking carcinoma, is a key or defining feature of several soft tissue tumors and may be seen in a wide variety of other tumors. This review will focus on those tumors defined at least in part by their epithelioid morphology, in particular epithelioid sarcoma, epithelioid malignant peripheral nerve sheath tumor, and sclerosing epithelioid fibrosarcoma. The role of loss of the SMARCB1 tumor-suppressor gene in the pathogenesis of these epithelioid soft tissue tumors will be discussed, as will their differential diagnosis with non-mesenchymal tumors, in particular carcinoma and melanoma.
Similar content being viewed by others


Epithelioid sarcoma
Epithelioid sarcoma (ES) is a distinctive sarcoma showing epithelial differentiation, most often involving the distal extremities of young patients. It was first formally described by Enzinger1 in 1970. It had previously been considered part of the spectrum of ‘tendosynovial sarcoma’, a term previously used to describe what we now consider to represent ES, clear cell sarcoma, and synovial sarcoma. Larger, more pleomorphic tumors, typically occurring in more proximal locations have been labeled ‘proximal-type’ ES. The morphological, immunohistochemical, and molecular genetics of ES shows considerable overlap with malignant extrarenal rhabdoid tumors (MERTs), as discussed below.
Clinical Features
Classical ES most commonly occur in adolescents and young adults, with a median age of 26 years. The tumor is approximately twice as common in males as in females. Classical ES most commonly involve the hands/fingers, followed by the wrist/lower arm and lower leg/knee but may occur in essentially any location.1, 2, 3 Involvement of tendons and aponeuroses is common but not invariably present. Most are small (3–6 cm) at the time of diagnosis. Proximal-type ES tends to occur in older adults, most often involving the deep soft tissues of the perineum, genital region, and pelvic soft tissues, and is present as much larger masses than do classical ES.4 Clinically, classical ES often presents as a small, indurated, sometime ulcerated nodule or nodules, frequently present for several weeks or longer, before coming to clinical attention. The clinical index of suspicion for a malignant lesion is often quite low.2 In contrast, proximal-type ES presents as a nonspecific soft tissue mass.3, 4
ES recurs in over 70% of cases, often as multiple subcutaneous nodules in the more proximal extremity. Nearly 50% of ES will eventually metastasize distantly, most often to the lymph nodes and the lungs but also to the skin and soft tissue sites.1, 2, 3 Metastatic ES is almost uniformly fatal. ES is not graded; adverse prognostic features are chiefly clinical and include male sex, proximal location, size >5 cm, and deep location. Proximal-type ES may metastasize earlier than classical variants.
Pathological Features
ES typically presents as relatively small (<5 cm), ulcerated, firm, single or multiple nodules, sometimes with overlying ulceration. Proximal-type ES presents as larger nonspecific soft tissue masses, often with grossly apparent areas of hemorrhage and necrosis. The morphological features of classical ES and some significant morphological variants are illustrated in Figures 1a–h. Morphological features of proximal-type ES are illustrated in Figures 2a–d. ES are often relatively circumscribed at low-power magnification but are invariably highly infiltrative tumors, frequently extending into the surrounding connective tissue in the form of small nests and single files of tumor cells.1, 2, 3 Tumor nodules often show central necrosis, mimicking necrobiotic granulomas. When individual tumor nodules may grow along tendons and fuse, they may produce a ‘garland-like’ appearance. The neoplastic cells may appear small and epithelioid, or may show greater pleomorphism and a rhabdoid appearance, particularly in larger, more proximally located tumors.3, 4 Although epithelioid morphology is always at least focally present, many ESs also show spindling, and this combination of features is characteristic. Rare ES consists chiefly of spindled cells.5 The nuclei of ES are relatively uniform appearing but show hyperchromatism, chromatin abnormalities, and irregular nuclear contours. Unusual morphologic changes include pseudovascular or pseudogland formation, myxoid change, calcification, and bone formation.6, 7 A mixed chronic inflammatory cell infiltrate is typically present and may occasionally obscure the underlying tumor.


At low-power magnification, epithelioid sarcomas often have a deceptively well-circumscribed growth pattern, mimicking some types of benign processes (a). However, infiltrative growth, often in the form of single cells and small tumor cell nests, is invariably present at the periphery of the tumor, likely accounting for the high rate of local recurrence (b). Central necrosis, a common feature of epithelioid sarcoma, may suggest the possibility of granulomatous disease (c). The cells of classical-type epithelioid sarcoma are typically small, uniform, and relatively bland (d). Epithelioid sarcomas typically show a mixture of epithelioid and more spindled forms; extensively spindled epithelioid sarcomas may mimic fibrous histiocytomas and other low-grade spindle cell tumors (e). Myxoid change in epithelioid sarcoma (f). Pseudogland formation in epithelioid sarcoma, mimicking adenocarcinoma. When these spaces are filled with blood, a vascular tumor is also a differential diagnostic consideration (g). Hyalinized collagen, calcification, and occasionally bone formation may be seen in some epithelioid sarcomas (h).

Proximal-type epithelioid sarcomas (PTESs) present as larger masses in the proximal extremities, often with large areas of necrosis (a). Stromal hyalinization in PTES (b). The cells of PTES show greater pleomorphism and more frequent mitotic figures than do those of the classical type (c). Rhabdoid morphology, with glassy intracytoplasmic inclusions, in PTES (d).
Immunohistochemical Features
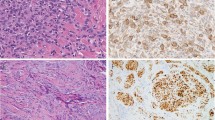
Using immunohistochemistry, ES expresses vimentin as well as a variety of different cytokeratins, including those of low and high molecular weights (Figure 3a).3, 4 They are generally negative, however, for cytokeratins 5/6.8 CD34 is expressed by ∼50% of cases, in contrast with <2% of carcinomas (Figure 3b).4, 9, 10 ES does not express other markers of endothelial differentiation, such as CD31, FLI-1/ERG, or vWF. Over 90% of ESs of all types show the loss of expression of the tumor-suppressor gene product SMARCB1/INI1/BAF47 (SMARCB1) (Figure 3c).11, 12, 13

Using immunohistochemistry, epithelioid sarcomas of all types typically show strong expression of cytokeratins, including both low and high molecular-weight forms (a). CD34 expression is seen in 50–60% of epithelioid sarcomas, in contrast to <2% of carcinomas (b). Complete loss of SMARCB1 (INI1) expression is seen in ∼90% of epithelioid sarcomas (c).
Genetic Features
Cytogenetic studies of ES show principally nonspecific chromosomal gains and losses.14, 15, 16, 17, 18, 19, 20 However, loss of 22q11, the location of the SMARCB1 gene, has been reported in few cases. Using FISH, SMARCB1 gene deletion has been reported in ES of both classical and proximal types. Mutation of SMARCB1, a feature of rhabdoid tumors, does not seem to occur in ES.7, 16, 21, 22
Differential Diagnosis
ES differs from granulomatous processes by virtue of its infiltrative growth pattern, the presence of true tumor cell necrosis as opposed to necrobiosis, and the presence of hyperchromatic cells showing expression of cytokeratins and loss of SMARCB1 expression. The loss of SMARCB1 expression is also helpful in the distinction of ES from carcinomas. Pseudovascular ES often expresses high molecular-weight cytokeratins, unlike endothelial cell neoplasms, and the lack of expression of CD31, FLI-1 protein, vWF, and SMARCB1. Spindled ES may be bland in appearance, closely mimicking cellular fibrous histiocytoma; cytokeratin immunostains may be extremely valuable here. Factor XIIIa immunostains are not helpful. Synovial sarcomas bear little if any resemblance to ES beyond cytokeratin expression.
Malignant rhabdoid tumors
Malignant rhabdoid tumor (MRT) of the kidney, initially described by Beckwith and Palmer23 in 1978 as a ‘rhabdomyosarcomatoid variant of Wilms’ tumor’ is now known to be unrelated to Wilms’ tumor.24, 25, 26 Most renal MRTs occur in children <1 year of age and have an aggressive clinical course, with rapid death from metastatic disease. MRT may also arise in the central nervous system, where they are referred to as ‘atypical teratoid/rhabdoid tumors’ and is in a disseminated form, lacking a clear dominant primary tumor.27, 28 Disseminated MRT invariably arises in infants, often as part of familial rhabdoid tumor kindreds.29
The great majority of soft tissue MRTs (MERT) occur in young children, and many cases reported as adult MERT more likely represent rhabdoid change within some other types of malignant neoplasms, so-called ‘composite rhabdoid tumor’ (CERT).25, 30 The term ‘MERT’ should be applied only to primitive malignant neoplasms showing at least in part rhabdoid morphology, almost always showing SMARCB1 protein loss and/or SMARC1 gene mutation (discussed below), and lacking any other line of differentiation.
Clinical Features
MERTs have been described in essentially any anatomical site but most frequently involve deep axial locations, such as the paraspinal region and the neck. The overwhelming majority of true MERTs occur in infants and young children, although rare bona fide cases in adults do occur. Similar to their renal counterparts, MERTs are characterized by aggressive clinical behavior, with fewer than 50% of patients surviving >5 years.24, 31, 32
Pathologic Features
MERT typically presents as soft, fleshy, gray to tan, hemorrhagic/necrotic masses, usually <5 cm in size. MERT is defined by the presence of ‘rhabdoid’ cells with eccentric vesicular nuclei, prominent nucleoli, and abundant cytoplasm-containing juxtanuclear eosinophilic, PAS-positive hyaline inclusions or globules (Figures 4a–c). Ultrastructurally, these inclusions represent whorls of intermediate filaments.33 Rhabdoid cells may be relatively rare in MERT, with some tumors consisting chiefly of a sheet-like proliferation of highly primitive-appearing round cells. Particularly in adults, morphologic features suggestive of PT–ES or some other definable primary neoplasms should be carefully searched for.

Malignant extrarenal rhabdoid tumor (MERT), consisting of a sheet-like proliferation of primitive round cells, occasionally with a small amount of eosinophilic cytoplasm (a). Rhabdoid inclusions may be only focally present in MERT, requiring careful high-power examination (b). Myxoid change in MERT may produce a cord or chain-like pattern of growth, mimicking other sarcomas such as extraskeletal myxoid chondrosarcoma (c). Loss of SMARCB1 expression, usually as the result of SMARCB1 mutation (as opposed to deletion) is the molecular hallmark of MERT (d). In contrast to epithelioid sarcoma, cytokeratin expression in MERT is typically much more limited in extent (e) and CD34 is often negative.
Immunohistochemical Findings
MERT typically expresses vimentin and is usually cytokeratin-positive, although this immunoreactivity is typically less pronounced than in PT–ES (Figures 4d and e). A wide variety of other markers may occasionally also be positive, including EMA, smooth muscle actin, CD99, CD57, synaptophysin, and S100 protein.34 SMARCB1 protein expression is consistently lost in true MERT, as compared with other malignant neoplasms showing rhabdoid morphology (Figures 5a–d) (CERT).13

In some instances, distinction of proximal-type epithelioid sarcoma from malignant extrarenal rhabdoid tumor is difficult and potentially subjective. This tumor occurred in the extremities of an older male and consisted of relatively uniform cells with numerous rhabdoid cytoplasmic inclusions (a). Cytokeratin expression, however, was entirely absent (b), whereas CD34 expression was present (c). Loss of SMARCB1 expression using immunohistochemistry (d) confirms that this tumor falls somewhere in the spectrum of epithelioid sarcoma and rhabdoid tumor. There are some data to suggest that the presence of SMARCB1 gene deletions (seen in epithelioid sarcoma) as opposed to gene mutations (seen in rhabdoid tumor) may distinguish these two entities.
Genetic Features
Cytogenetic studies have consistently identified 22q aberrations including monosomy of chromosome 22 with or without partial deletion of the remaining chromosome 22 in MERT.35, 36 It has subsequently been shown that the vast majority of MRTs in all locations show deletions or mutations of the SMARCB1 gene.13, 37, 38, 39, 40, 41
SMARCB1 (INI1, SNF5, BAF47)
SMARCB1 is a member of the SWI/SNF chromatin-remodeling complex and is thought to have a direct role in activation and suppression of gene expression by displacing DNA from histones and allowing transcription.39, 42 Germline mutations of the SMARCB1 gene are the causes of the rhabdoid predisposition syndrome.39, 42 With only very rare exceptions, CERT lacks SMARCB1 alterations.43, 44 Alterations of the SMARCB1 gene affect multiple pathways, which may be central to the pathogenesis of this tumor including the p16/INK4A, p14/ARF, and p21 (CIP1/WAF1) pathways.13, 39, 42 As noted above, the loss of SMARCB1 expression is also seen in >90% of ESs of all types. ESs appear to differ from MERT in that they typically show deletions of SMARCB1, rather than mutations.22, 37, 38, 45, 46 The loss of SMARCB1 expression is also seen in essentially all renal medullary carcinomas and in subsets of epithelioid malignant peripheral nerve sheath tumors (EMPNSTs), myoepithelial carcinomas, and extraskeletal myxoid chondrosarcomas.13 Germline SMARCB1 alterations also appear to underlie schwannomatosis.47, 48, 49 Very rare instances of MRT and epithelioid MPNST arising in schwannomatosis have been reported, presumably reflecting second somatic mutations.50, 51
Epithelioid malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumors are relatively rare, accounting for roughly 5% of all soft tissue sarcomas.52, 53 Approximately one-third occur in association with neurofibromatosis type 1. EMPNST accounts for <5% of all MPNSTs.
Clinical Features
As with typical spindle cell MPNST, EMPNST occurs most often in patients 20–50 years of age, with a slight male predominance. Most involve major nerves,54 although rare tumors may occur in unusual locations such as the skin.55 For unclear reasons, EMPNST accounts for the great majority of MPNST that arises within schwannomas (an exceedingly rare event).51, 56 EMPNSTs are aggressive lesions, with distant metastases eventually occurring in ∼50% of patients, typically to the lungs.54
Pathological Features
Selected morphological features of EMPNST are illustrated in Figures 6a–f). By definition, EMPNST is composed entirely or nearly entirely of epithelioid cells, resembling those seen in melanoma. The tumors are typically relatively well-circumscribed but microscopically infiltrative and show a variably hyalinized collagenous stroma. The neoplastic cells typically grow in a distinctly nested pattern, at least in part, with other areas usually showing a cord-like pattern of growth. Variable stromal myxoid change may be seen. Metaplastic bone formation is seen on occasion, as is clear cell or signet-ring change. Rhabdoid inclusions may be present. Mitotic activity is invariably present and necrosis is frequently seen.

Cutaneous malignant melanoma (a), showing striking rhabdoid morphology (composite rhabdoid tumor) (b). Retained expression of SMARCB1 is usually seen in composite rhabdoid tumors (that is, other neoplastic types with rhabdoid morphology) (c). With increased use of SMARCB1 immunohistochemistry, however, it has become apparent that some ‘true’ composite rhabdoid tumors exist, such as this rhabdomyosarcoma with rhabdoid-type cytoplasmic inclusions (d), which showed diffuse expression of desmin (e) and MyoD1 (e, inset) but lacked SMARCB1 expression (f). Non-neoplastic histiocytes serve as an internal control for SMARCB1.
Immunohistochemical Features
EMPNST is almost always strongly and diffusely positive for the S100 protein, in contrast to other types of MPNSTs, which typically show only patchy and weak expression (Figure 7g). EMPNST characteristically shows abundant collagen IV expression surrounding nests of cells (Figure 7h). Unlike melanomas and clear cell sarcomas, which also show diffuse S100 protein expression, EMPNST is negative for melanocytic markers such as HMB45, Melan-A, and tyrosinase. Approximately 50% of EMPNSTs show loss of SMARCB1 expression, a finding that may be of value in their differential diagnosis with melanoma and clear cell sarcoma, both of which show retained expression of this protein.13


Epithelioid malignant peripheral nerve sheath tumor (EMPNST) presenting as a well-circumscribed but non-encapsulated soft tissue mass (a). At higher power magnification, EMPNSTs are typified by a distinctly nested pattern of growth and by ‘melanoma-like’ epithelioid cells with prominent macronucleoli (b). Unlike other forms of MPNST, EMPNST not uncommonly occurs in superficial locations, potentially mimicking cutaneous melanoma (c). Note, however, the absence of a junctional component and superficial dermal involvement. Most EMPSTs are obviously malignant, with high-grade cytology and tumor cell necrosis (d). Some EMPSTs may mimic a benign peripheral nerve sheath tumor, as in this pseudo-encapsulated with cellular and myxoid zones, simulating the Antoni A and B patterns of schwannoma (e). At higher power magnification, however, prominent nucleoli and frequent mitotic figures were present, diagnostic of EMPNST (f). Using immunohistochemistry, EMPNST typically shows strong, diffuse S100 protein expression (g) but lacks the expression of specific melanocytic markers such as Melan-A. The presence of abundant collagen IV production around nests of cells is also characteristic of EMPNST (h).
Differential Diagnosis
EMPNSTs are most often confused with epithelioid schwannomas, conventional melanomas, and clear cell sarcomas, particularly as all of these tumors typically show diffuse S100 protein expression. Epithelioid schwannomas tend to be smaller than EMPNST, are more often superficial in location, show a well-formed capsule, and are composed of very bland, small epithelioid cells, lacking mitotic activity or necrosis. Conventional malignant melanomas may very closely resemble EMPNST but express HMB45, Melan-A, and/or tyrosinase in roughly 80% of cases. Melanoma shows retained expression of the SMARCB1 protein, in contrast to many EMPNSTs. Clear cell sarcoma usually shows a greater degree of cellular spindling than do EMPNST, often contains Touton-like neoplastic giant cells, and expresses melanocytic markers. Clear cell sarcomas also show t(12;22) (EWSR1-ATF1) in the great majority of cases;57 this translocation is not seen in EMPNST.
Sclerosing epithelioid fibrosarcoma
Sclerosing epithelioid fibrosarcoma (SEF) is a relatively newly described malignant fibroblastic tumor, considered by the current WHO classification of soft tissue and bone tumors to represent a distinct variant of fibrosarcoma.58 However, there is considerable morphological and genetic data to suggest a link between SEF and low-grade fibromyxoid sarcoma (LGFMS) (see below).59, 60, 61, 62, 63
Clinical Features
SEF occurs over a wide age range in patients of either sex as a deeply situated, often large soft tissue mass, typically in the lower extremities.59, 60, 61, 62, 63, 64 SEF is generally considered a low-grade sarcoma, although a more aggressive clinical course has been suggested in one relatively large series.63 SEF metastasizes to the lungs with overall mortality of 25–57%.63, 64
Pathological Features
SEFs are usually grossly well-circumscribed, ranging from 5 to 10 cm in size. Microscopically, they show infiltrative borders and are composed of cords, chains, and nests of moderately pleomorphic epithelioid cells, often with clear cytoplasm, embedded in a densely hyalinized stroma (Figures 8a–d). Many cases also contain small spindled foci, resembling adult fibrosarcoma65 or LGFMS (Figure 8e).66 Mitotic activity is typically low and necrosis is not usually present. Metaplastic bone formation may be present.


Sclerosing epithelioid fibrosarcoma (SEF) showing areas of both conventional spindled fibrosarcoma (top) and epithelioid morphology (bottom) (a). Spindled and epithelioid morphologies in SEF (b). Epithelioid cells with pericellular clearing, growing in a densely hyalinized matrix in SEF (c). The epithelioid fibroblastic cells of SEF tend to be relatively uniform, with infrequent mitotic figures (d). Hybrid SEF-low-grade fibromyxoid sarcoma (LGFMS) with hyalinized, epithelioid foci juxtaposed to myxoid, spindled nodules (e). Most LGFMS and hybrid SEF–LGFMS show FUS–CREB3L2 fusions, as do a smaller percentage of pure SEF. Rarely, however, these tumors may show EWSR1 rather than FUS rearrangements, as in this SEF (f) positive for EWSR1 rearrangement using FISH (g).
Immunohistochemical Features
SEF typically shows a fibroblastic phenotype, with expression chiefly of vimentin. Very recently, expression of the MUC4 glycoprotein has been described as a sensitive marker of SEF, both ‘pure’ SEF and those showing hybrid features of LGFMS.67 Epithelial marker expression has been reported in some SEFs64 but seems to be very uncommon, in my experience.
Genetic Features and Relationship to LGFMS
The possibility that SEF might not represent a single histopathological entity was first raised by Eyden et al68 in a morphologic, immunohistochemical, and ultrastructural study of five cases. A possible link between SEF and LGFMS was first explicitly suggested by Antonescu et al,63 who noted the presence of hypocellular collagenous zones and juxtaposed myxoid nodules, identical to LGFMS, in 4 of 16 studied SEF. Additional evidence linking SEF and LGFMS comes from two genetic studies of LGFMS, from Guillou et al62 and Reid et al69 Guillou et al62 studied 63 tumors with morphological features of LGFMS and 66 non-LGFMS for the LGFMS-specific translocation t (7;16)(q32-34;p11)(FUS–CREB3L2/L1). Positive results were found in 81% of putative LGFMS and only 7 of 52 (13%) controls; of these seven positive non-LGFMS, four had been previously labeled ‘SEF’. Along similar lines, one of the 4 genetically confirmed LGFMS reported by Reid et al69 showed at least in part classical features of SEF. Rehki et al70 also reported a hybrid SEF-LGMFS showing FUS rearrangement.70
Two very recent studies have more specifically examined the relationship between SEF and LGFMS. Wang et al71 studied a group of 22 ‘pure’ SEF specifically lacking LGFMS-like areas and found FUS rearrangements in only two cases. Doyle et al67 identified FUS rearrangement in 6/23 ‘pure’ SEF, one of which was shown to also have CREB3L2 rearrangement and in two of the six hybrid SEF–LGFMS. Interestingly, two other hybrid tumors showed EWSR, rather than FUS rearrangement (Figure 8g).
It would thus appear that at least some cases of ‘SEF’ represent instead morphological variants of LGFMS, in particular those cases that show hybrid morphological features. Additionally, subsets of pure SEF show FUS and CREB3L2 rearrangements, suggesting that they too are closely related (if not identical) to LGFMS. It is unclear whether FUS-negative SEF represents an entity distinct from LGFMS, or perhaps a form of morphological progression in a heterogeneous group of soft tissue tumors. In this respect, it should be noted that areas identical to SEF may be seen in sclerosing rhabdomyosarcomas,72 ossifying fibromyxoid tumors,73 malignant peripheral nerve sheath tumors, hyalinizing leiomyosarcomas, and osteosarcomas, in our experience.
Other sarcomas with epithelioid features
Epithelioid variants of many different soft tissue tumors, including angiosarcoma,74 leiomyosarcoma,75 myxofibrosarcoma,76 pleomorphic liposarcoma,77 mammary myofibroblastoma,78 rhabdomyosarcoma,79 and inflammatory myofibroblastic tumor80 have been reported. The chief significance of these unusual morphological variants is in their potential mimicry of true epithelioid tumors, in particular carcinoma (Figures 9a–h). The reader is referred to the references for additional information about these rare lesions.


Epithelioid pleomorphic liposarcoma mimicking adrenal cortical carcinoma (a). The presence of pleomorphic lipoblasts confirms the correct diagnosis (b). Epithelioid change (c) in myxofibrosarcoma (d) may mimic carcinoma or a malignant myoepithelial tumor. Epithelioid mammary myofibroblastoma may closely simulate lobular carcinoma (e). Careful examination of such lesions typically discloses more typical areas of myofibroblastoma, with small aggregates of bland-spindled cells and wiry collagen (f). Epithelioid rhabdomyosarcoma (g), confirmed with positive myogenin immunohistochemistry (h). These extremely rare lesions may closely simulate carcinoma and require an extended panel of immunostains for correct diagnosis.
Conclusion
Although epithelioid change may be seen in a wide variety of soft tissue tumors, this morphology is most characteristic of ES, MERT, EMPNST, and SEF. The precise relationship between SMARCB1 abnormalities and epithelioid morphology remains to be fully elucidated; however, the presence of such aberrations in ES, MERT, and EMPNST suggests that this is more than a coincidence. From a practical perspective, improved recognition of the various sarcomas that may show epithelioid morphology is critical for their distinction from other epithelioid malignancies and for correct treatment.
References
Enzinger FM . Epithelioid sarcoma: a sarcoma simulating a granuloma or a carcinoma. Cancer 1970;26:1029–1041.
Chase DR, Enzinger FM . Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am Jl Surg Pathol 1985;9:241–263.
Chbani L, Guillou L, Terrier P et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 2009;131:222–227.
Guillou L, Wadden C, Coindre JM et al. ‘Proximal-type’ epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol 1997;21:130–146.
Mirra JM, Kessler S, Bhuta S et al. The fibroma-like variant of epithelioid sarcoma. A fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer 1992;69:1382–1395.
Koplin SA, Nielsen GP, Hornicek FJ et al. Epithelioid sarcoma with heterotopic bone: a morphologic review of 4 cases. Int J Surg Pathol 2010;18:207–212.
Raoux D, Peoc'h M, Pedeutour F et al. Primary epithelioid sarcoma of bone: report of a unique case, with immunohistochemical and fluorescent in situ hybridization confirmation of INI1 deletion. Am J Surg Pathol 2009;33:954–958.
Lin L, Skacel M, Sigel JE et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol 2003;30:114–117.
Laskin WB, Miettinen M . Epithelioid sarcoma: new insights based on an extended immunohistochemical analysis. Arch Pathol Lab Med 2003;127:1161–1168.
Miettinen M, Fanburg-Smith JC, Virolainen M et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol 1999;30:934–942.
Orrock JM, Abbott JJ, Gibson LE et al. INI1 and GLUT-1 expression in epithelioid sarcoma and its cutaneous neoplastic and nonneoplastic mimics. Am J Dermatopathol 2009;31:152–156.
Hornick JL, Dal Cin P, Fletcher CD . Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 2009;33:542–550.
Hollmann TJ, Hornick JL . INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol 2011;35:e47–e63.
Stenman G, Kindblom LG, Willems J et al. A cell culture, chromosomal and quantitative DNA analysis of a metastatic epithelioid sarcoma. Deletion 1p, a possible primary chromosomal abnormality in epithelioid sarcoma. Cancer 1990;65:2006–2013.
Reeves BR, Fisher C, Smith S et al. Ultrastructural, immunocytochemical, and cytogenetic characterization of a human epithelioid sarcoma cell line (RM-HS1). JNCI 1987;78:7–18.
Modena P, Lualdi E, Facchinetti F et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 2005;65:4012–4019.
Iwasaki H, Ohjimi Y, Ishiguro M et al. Epithelioid sarcoma with an 18q aberration. Cancer Genet Cytogenet 1996;91:46–52.
Feely MG, Fidler ME, Nelson M et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet 2000;119:155–157.
Cordoba JC, Parham DM, Meyer WH et al. A new cytogenetic finding in an epithelioid sarcoma, t(8;22)(q22;q11). Cancer Genet Cytogenet 1994;72:151–154.
Brassesco MS, Valera ET, Castro-Gamero AM et al. Cytogenetic findings in an epithelioid sarcoma with angiomatoid features. A case report. Genet Mol Res 2009;8:1211–1217.
Kosemehmetoglu K, Kaygusuz G, Bahrami A et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol 2011;35:891–897.
Kohashi K, Izumi T, Oda Y et al. Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol 2009;40:349–355.
Beckwith JB, Palmer NF . Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer 1978;41:1937–1948.
Weeks DA, Beckwith JB, Mierau GW et al. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol 1989;13:439–458.
Wick MR, Ritter JH, Dehner LP . Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol 1995;12:233–248.
Vujanic GM, Sandstedt B, Harms D et al. Rhabdoid tumour of the kidney: a clinicopathological study of 22 patients from the International Society of Paediatric Oncology (SIOP) nephroblastoma file. Histopathology 1996;28:333–340.
Biegel JA . Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 2006;20:E11.
White FV, Dehner LP, Belchis DA et al. Congenital disseminated malignant rhabdoid tumor: a distinct clinicopathologic entity demonstrating abnormalities of chromosome 22q11. Am J Surg Pathol 1999;23:249–256.
Lee HY, Yoon CS, Sevenet N et al. Rhabdoid tumor of the kidney is a component of the rhabdoid predisposition syndrome. Pediatr Dev Pathol 2002;5:395–399.
Ogino S, Ro TY, Redline RW . Malignant rhabdoid tumor: A phenotype? An entity?--A controversy revisited. Adv Anat Pathol 2000;7:181–190.
Beckwith JB . Wilms' tumor and other renal tumors of childhood: a selective review from the National Wilms' Tumor Study Pathology Center. Hum Pathol 1983;14:481–492.
Tsokos M, Kouraklis G, Chandra RS et al. Malignant rhabdoid tumor of the kidney and soft tissues. Evidence for a diverse morphological and immunocytochemical phenotype. Arch Path Lab Med 1989;113:115–120.
Haas JE, Palmer NF, Weinberg AG et al. Ultrastructure of malignant rhabdoid tumor of the kidney. A distinctive renal tumor of children. Hum Pathol 1981;12:646–657.
Fanburg-Smith JC, Hengge M, Hengge UR et al. Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immunohistochemical study of 18 cases. Ann Diagn Pathol 1998;2:351–362.
Shashi V, Lovell MA, von Kap-herr C et al. Malignant rhabdoid tumor of the kidney: involvement of chromosome 22. Genes Chromosomes Cancer 1994;10:49–54.
Versteege I, Sevenet N, Lange J et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998;394:203–206.
Eaton KW, Tooke LS, Wainwright LM et al. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 2011;56:7–15.
Bruggers CS, Bleyl SB, Pysher T et al. Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatric Blood Cancer 2011;56:1026–1031.
Roberts CW, Biegel JA . The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther 2009;8:412–416.
Bourdeaut F, Freneaux P, Thuille B et al. hSNF5/INI1-deficient tumours and rhabdoid tumours are convergent but not fully overlapping entities. J Pathol 2007;211:323–330.
Sigauke E, Rakheja D, Maddox DL et al. Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod Pathol 2006;19:717–725.
Biegel JA, Kalpana G, Knudsen ES et al. The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 2002;62:323–328.
Perry A, Fuller CE, Judkins AR et al. INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol 2005;18:951–958.
Donner LR, Wainwright LM, Zhang F et al. Mutation of the INI1 gene in composite rhabdoid tumor of the endometrium. Hum Pathol 2007;38:935–939.
Sullivan LM, Folpe AL, Pawel BR et al. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol 2012;26:385–392.
Jackson EM, Sievert AJ, Gai X et al. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res 2009;15:1923–1930.
Hulsebos TJ, Kenter SB, Jakobs ME et al. SMARCB1/INI1 maternal germ line mosaicism in schwannomatosis. Clin Genet 2010;77:86–91.
Bacci C, Sestini R, Provenzano A et al. Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics 2010;11:73–80.
Smith MJ, Boyd CD, MacCollin MM et al. Identity analysis of schwannomatosis kindreds with recurrent constitutional SMARCB1 (INI1) alterations. Clin Genet 2009;75:501–502.
Swensen JJ, Keyser J, Coffin CM et al. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet 2009;46:68–72.
Carter JM, O'Hara C, Dundas G et al. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with ‘neuroblastoma-like’ schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol 2012;36:154–160.
Fletcher CDM, Unni KK, Mertens F World Health O, International Agency for Research on C. Pathology and Genetics of Tumours of Soft Tissue and Bone Vol. IARC Press: Lyon, 2002.
Weiss SW, Goldblum JR . Enzinger & Weiss's Soft Tissue Tumors Vol. 5th edn Mosby, Elsevier: Philadelphia, 2008.
Laskin WB, Weiss SW, Bratthauer GL . Epithelioid variant of malignant peripheral nerve sheath tumor (malignant epithelioid schwannoma). Am J Surg Pathol 1991;15:1136–1145.
Allison KH, Patel RM, Goldblum JR et al. Superficial malignant peripheral nerve sheath tumor: a rare and challenging diagnosis. Am J Clin Pathol 2005;124:685–692.
McMenamin ME, Fletcher CD . Expanding the spectrum of malignant change in schwannomas: epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: a study of 17 cases. Am J Surg Pathol 2001;25:13–25.
Kosemehmetoglu K, Folpe AL . Clear cell sarcoma of tendons and aponeuroses, and osteoclast-rich tumour of the gastrointestinal tract with features resembling clear cell sarcoma of soft parts: a review and update. J Clin Pathol 2010;63:416–423.
Fletcher CDM, Bridge JA, Hogendoorn PCW . WHO Classification of Tumours of Soft Tissue and Bone Vol. 4th edn IARC Press: Lyons, 2013.
Doyle LA, Wang WL, Dal Cin P et al. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: association with FUS gene rearrangement. Am J Surg Pathol 2012;36:1444–1451.
Rekhi B, Deshmukh M, Jambhekar NA . Low-grade fibromyxoid sarcoma: a clinicopathologic study of 18 cases, including histopathologic relationship with sclerosing epithelioid fibrosarcoma in a subset of cases. Ann Diagn Pathol 2011;15:303–311.
Evans HL . Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 2011;35:1450–1462.
Guillou L, Benhattar J, Gengler C et al. Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol 2007;31:1387–1402.
Antonescu CR, Rosenblum MK, Pereira P et al. Sclerosing epithelioid fibrosarcoma: a study of 16 cases and confirmation of a clinicopathologically distinct tumor. Am J Surg Pathol 2001;25:699–709.
Meis-Kindblom JM, Kindblom LG, Enzinger FM . Sclerosing epithelioid fibrosarcoma. A variant of fibrosarcoma simulating carcinoma. Am J Surg Pathol 1995;19:979–993.
Bahrami A, Folpe AL . Adult-type fibrosarcoma: a reevaluation of 163 putative cases diagnosed at a single institution over a 48-year period. Am J Surg Pathol 2010;34:1504–1513.
Evans HL . Low-grade fibromyxoid sarcoma. A report of 12 cases. Am J Surg Pathol 1993;17:595–600.
Doyle LA, Moller E, Dal Cin P et al. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol 2011;35:733–741.
Eyden BP, Manson C, Banerjee SS et al. Sclerosing epithelioid fibrosarcoma: a study of five cases emphasizing diagnostic criteria. Histopathology 1998;33:354–360.
Reid R, Barrett A, Hamblen DL . Sclerosing epithelioid fibrosarcoma. Histopathology 1996;28:451–455.
Rekhi B, Folpe AL, Deshmukh M et al. Sclerosing epithelioid fibrosarcoma—a report of two cases with cytogenetic analysis of FUS gene rearrangement by FISH technique. Pathol Oncol Res 2011;17:145–148.
Wang WL, Evans HL, Meis JM et al. FUS rearrangements are rare in 'pure' sclerosing epithelioid fibrosarcoma. Mod Pathol 2012;25:846–853.
Folpe AL, McKenney JK, Bridge JA et al. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol 2002;26:1175–1183.
Folpe AL, Weiss SW . Ossifying fibromyxoid tumor of soft parts: a clinicopathologic study of 70 cases with emphasis on atypical and malignant variants. Am J Surg Pathol 2003;27:421–431.
Fletcher CD, Beham A, Bekir S et al. Epithelioid angiosarcoma of deep soft tissue: a distinctive tumor readily mistaken for an epithelial neoplasm. Am J Surg Pathol 1991;15:915–924.
de Saint Aubain Somerhausen N, Fletcher CD . Leiomyosarcoma of soft tissue in children: clinicopathologic analysis of 20 cases. Am J Surg Pathol 1999;23:755–763.
Nascimento AF, Bertoni F, Fletcher CD . Epithelioid variant of myxofibrosarcoma: expanding the clinicomorphologic spectrum of myxofibrosarcoma in a series of 17 cases. Am J Surg Pathol 2007;31:99–105.
Miettinen M, Enzinger FM . Epithelioid variant of pleomorphic liposarcoma: a study of 12 cases of a distinctive variant of high-grade liposarcoma. Mod Pathol 1999;12:722–728.
Magro G . Epithelioid-cell myofibroblastoma of the breast: expanding the morphologic spectrum. Am J Surg Pathol 2009;33:1085–1092.
Jo VY, Marino-Enriquez A, Fletcher CD . Epithelioid rhabdomyosarcoma: clinicopathologic analysis of 16 cases of a morphologically distinct variant of rhabdomyosarcoma. Am J Surg Pathol 2011;35:1523–1530.
Gleason BC, Hornick JL . Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol 2008;61:428–437.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Folpe, A. Selected topics in the pathology of epithelioid soft tissue tumors. Mod Pathol 27 (Suppl 1), S64–S79 (2014). https://doi.org/10.1038/modpathol.2013.175
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.175
Keywords
This article is cited by
-
Epithelioid Sarcoma of the External Auditory Canal: An Uncommon Tumor at an Unusual Site and a Brief Overview of the Literature
Head and Neck Pathology (2021)
-
Myoepithelioma-like tumor of the vulvar region: a case report in China and review of the literature
Diagnostic Pathology (2020)
-
Variable Expression of S100 Protein in Sinonasal Malignant Mucosal Melanoma: A Potential Diagnostic Pitfall
Head and Neck Pathology (2020)
-
What is new in epithelioid soft tissue tumors?
Virchows Archiv (2020)
-
Sarcoma in neurofibromatosis 2: case report and review of the literature
Familial Cancer (2019)
