
Abstract
Histiocytic sarcoma (HS) is a rare but controversial hematopoietic neoplasm. In the past, malignancies have been misclassified as histiocytic tumors due to overlapping histologic features and inadequate phenotypic data. CD163, a recently characterized hemoglobin scavenger receptor, appears to be a ‘specific’ marker of histiocytic lineage and a promising diagnostic tool for evaluating histiocytic neoplasms. Five cases of HS were studied to further elucidate the clinicopathologic features of these rare tumors and to demonstrate the diagnostic utility of CD163. Criteria for diagnosis included histologic and immunohistochemical evidence of histiocytic differentiation, CD45 positivity, and exclusion of lymphoid, epithelial, melanocytic and dendritic cell phenotype. Sites of disease included the colon (two cases), palate, inguinal lymph node, and testis. The clinical course was aggressive in 4/5 patients (survival=2–15 months). One patient with localized disease of the palate, survived 17 years after diagnosis. All patients with poor survival had tumors ≥3.5 cm. Histologically, all cases showed diffuse architecture with large, discohesive polygonal cells. Spindling of cells was focally noted. Hemophagocytosis was identified in 3/5 cases. A prominent inflammatory background was present in 4/5 tumors. All cases were immunoreactive for CD45, CD163, CD68, and lysozyme. S-100 was focally positive in 4/5 cases. Antibodies for melanocytic, epithelial, lymphoid, and dendritic cell markers were negative. Molecular studies showed monoclonal IgH gene rearrangements in three cases. Our findings suggest that HS is an uncommon neoplasm frequently extranodal in presentation and aggressive in behavior, with rare exceptions. Stage of disease and possibly tumor size are significant prognostic indicators. Molecular studies remain controversial in the diagnosis. The morphologic and phenotypic features are relatively uniform; however, the diagnosis requires exclusion of more common neoplasms by extensive immunophenotypic studies. CD163 appears to be a specific histiocytic marker and is important in establishing the diagnosis of HS.
Similar content being viewed by others

Main
Histiocytic sarcoma (HS), formerly designated as true histiocytic lymphoma, consists of exceedingly rare hematopoietic neoplasms, representing <1% of all non-Hodgkin's lymphomas.1, 2 Many inconsistencies in the terminology and diagnostic criteria of these lesions have historically complicated their recognition and characterization. In fact, neoplasms originally classified as ‘reticulum cell sarcomas’3 and later ‘histiocytic lymphomas’ by Rappaport4 have encompassed a biologically heterogeneous group of disorders, the majority of which are now known to be high-grade T- or B-cell, non-Hodgkin lymphomas.5, 6, 7, 8, 9, 10
The term ‘histiocytic sarcoma’ was introduced in 1970 by Mathé et al;11 however, this and previous descriptions were based strictly on the histologic similarities of the cells to macrophages. Since these original papers, increasing emphasis has been placed on cytochemical and immunohistochemical characterization of these neoplasms in an attempt to establish true histiocytic lineage.12, 13, 14, 15, 16, 17 This diagnostic challenge is further complicated as it is necessary to distinguish HS from other histiocytic processes, both benign and malignant, to include the hemophagocytic syndrome,18, 19, 20 malignant histiocytosis,10, 18, 19, 20, 21 and monocytic leukemia.17, 19
The diagnosis of HS relies predominantly on the verification of histiocytic lineage and the exclusion of other, poorly differentiated, large cell malignancies (ie lymphoma, carcinoma, melanoma) by way of extensive immunophenotypic investigation. Fulfilling these conditions, particularly establishing histiocytic differentiation, has been problematic due to a high incidence of cross-reactivity of many immunohistochemical antibodies originally believed to be specific for histiocytes.22, 23, 24 The recent characterization of CD163,25, 26 a hemoglobin scavenger receptor protein, has offered a means of identifying histiocytic cells with a greater degree of specificity and is a promising marker in the diagnosis of true histiocytic malignancies. Finally, the contribution of molecular gene rearrangement studies as a diagnostic tool for histiocytic tumors remains unclear.14, 27, 28, 29
At the present time, the World Health Organization defines HS as a malignancy with morphologic and immunophenotypic features that resemble those of mature tissue histiocytes.2 Despite the application of more stringent diagnostic criteria; however, an increasing number of reports continue to shed light on these uncommon malignancies.9, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44 Extranodal presentation is frequent and the clinical course is generally aggressive. Unfortunately, persistent clinical and histopathologic disparities in these studies, in addition to the overall rarity of these neoplasms, have continued to obscure this diagnosis and prevented a full appreciation of their clinical behavior. We report the clinicopathologic features of five cases of HS to further elucidate the intriguing nature of these rare malignancies and introduce the application of a novel immunohistochemical marker, CD163, to attempt to verify the exact lineage of these neoplasms.
Materials and methods
Specimens and Selection Criteria
Five cases of HS were selected from 19 that were originally diagnosed as ‘histiocytic sarcoma,’ ‘true histiocytic lymphoma’ or ‘interdigitating dendritic cell/histiocytic sarcoma’ from the files of the Armed Forces Institute of Pathology between 1985 and 2001. Most of the excluded cases were reclassified following immunohistochemical analysis. Three cases were excluded at the onset of the study because they lacked paraffin blocks for ancillary studies. Case inclusion required morphologic and immunophenotypic features consistent with histiocytic lineage (CD68, CD163, lysozyme, MAC387 reactivity) and the absence of immunoreactivity for an extensive panel of lymphoid, accessory/dendritic cell, epithelial, and melanocytic markers. Cases not meeting these criteria, and those that lacked CD45 (LCA) expression, were excluded. In addition, patients with a leukemic or widely disseminated initial presentation were not considered in the present study.
Clinical History
Medical records were retrieved for all cases in this study. Available records were reviewed for clinical presentation, tumor size, stage, treatment, and clinical outcome.
Histology and Immunohistochemistry
Hematoxylin–eosin slides were reviewed in all cases. An extensive panel of immunohistochemical antibodies was applied to 4 μm, formalin-fixed, paraffin-embedded tissue sections according to standard methods. A list of the primary antibodies, clones, vendors, pretreatment regimens, and working dilutions used in this study are provided in Table 1. The sections were deparaffinized and treated with the specified antigen retrieval method, followed by overnight incubation with the primary antibodies at room temperature. Endogenous enzyme activity was quenched by treating the tissue with 3% hydrogen peroxide for 10 min. A streptavidin–biotin–peroxidase system (Vectastain Elite® ABC Kit, Vector Laboratories, Burlingame, CA, USA) with a diaminobenzidine (DAB) chromogen was then used to develop the stain. Sections were counterstained with Gill's hematoxylin, dehydrated in ethanol and cleared in xylene. Appropriate positive and negative controls were used for all antibodies.
Immunohistochemical stains were interpreted as positive if at least 10% of the neoplastic cells showed intense immunoreactivity. Specimens were considered focally positive if immunoreactivity was identified within the cells of interest but comprised less than 10% of the tumor. Cell proliferation was examined by Ki-67 antibody staining.
Gene Rearrangement Studies
Molecular diagnostic studies were performed by polymerase chain reaction (PCR) for the immunoglobulin heavy chain (IgH), T-cell receptor β (TCRβ) and T-cell receptor γ (TCRγ) genes, as previously described.45, 46, 47, 48 DNA was extracted from formalin-fixed, paraffin-embedded tissues in all cases and purified according to previously published methods.46, 48 Consensus primers were used for detection of IgH chain (FR3A V region and CFW1 J region), TCRβ chain (VJ1, VJ2, D1J2, and D2J2), and TCRγ chain (V2, V3, V4, V8, V9, JGT3, JGT4, and JGT12) gene rearrangements. PCR analyses were performed using a Perkin–Elmer Thermal Cycler 9600 (Applied Biosystems, Foster City, CA, USA) for 40 cycles using the following parameters: initial 10 min incubation at 94°C, denaturation for 1 min at 94°C, annealing for 1 min at 52°C, elongation for 1 min at 72°C, and a final elongation step for an additional 5 min at 72°C. All reactions were performed in duplicate along with positive and negative control specimens. The positive controls consisted of monoclonal cell lines (SUD 5, QA2#3, and Sup T-1 for IgH, TCRβ and TCRγ, respectively) and the negative control specimens were benign tonsil tissue and sterile water. The products underwent separation and detection by capillary electrophoresis using an ABI Prism® 310 Genetic Analyzer (Applied Biosystems). The resultant banding patterns were analyzed for the presence of monoclonality.
Cases were reported as monoclonal if a dominant peak was observed on the electropherogram and was reproducible on duplicate tests (±1 base pair) using the same specimen and primer set. A peak was considered dominant only when the amplitude was greater than all peaks present and was at least twice that of the local background. Reactions were determined to be negative (ie polyclonal) if multiple peaks were present but no dominant peak was detected or, in cases in which lymphocytes were sparse, a dominant peak was not reproducible on duplicate tests. Cases were considered not amplifiable (ie no PCR products were obtained) if the tracing was essentially the same as the water control.
Electron Microscopy
Representative biopsy samples from three cases were analyzed by electron microscopy. One case was extracted from the paraffin tissue block, deparaffinized with xylene and postfixed in glutaraldehyde. The remaining two cases were primarily fixed in glutaraldehyde and processed by conventional methods.49 Sections (1 μm) were stained with toluidine blue for orientation. Thin sections were then cut on an ultramicrotome, stained with uranyl acetate/lead citrate, and examined in an electron microscope. Numerous fields were studied to assess cell membranes and to look for various cytoplasmic constituents including lysosomes and Birbeck granules.
Results
Five out of the original 19 cases in this study met the criteria for HS. Of the excluded cases, nine could be reclassified, after an extensive immunohistochemical work-up, as anaplastic large cell lymphoma (three cases), diffuse large B-cell lymphoma (three cases), interdigitating dendritic cell sarcoma/tumor (two cases) and myeloid sarcoma (one case). The remaining two tumors could not be categorized, demonstrating no specific immunophenotype, in addition to lacking CD45 immunoreactivity. As mentioned previously, further studies were not possible for three/19 specimens due to unavailable tissue blocks.
Clinical Findings
Available clinical data including clinical presentation, tumor size, stage, disease course, and clinical outcome are summarized in Table 2. There were three males and two females (M:F=3:2) with ages ranging from 20 to 73 years (mean, 57.8 years). One case presented in the inguinal lymph node and four cases presented in extranodal sites including the colon (two cases), testis (one case), and palate (one case). Three patients were found to have stage IV disease on further work-up defined by involvement of multiple extralymphatic organs (Case 2), bone marrow (Case 3), or liver (Case 4). The colon was involved in each of these cases resulting in abdominal pain as one of the presenting complaints. Bone marrow tissue was not available for histologic review. By report, none of the patients manifested a leukemic component, including those with high-stage disease.
Follow-up information (Table 2) was obtained in all cases and ranged from 2 months to 17 years. Although the number of patients in this study was small, the stage of disease appeared to correlate with the clinical course. The average length of survival among the three patients with known stage IV disease was 7 months. In contrast, Patient 1, who presented with localized extranodal disease involving the palate (stage IE), remained disease free following his initial treatment and died of unrelated causes, 17 years after his initial diagnosis.
Tumor size also appeared to be an important predictor of clinical behavior. Each case that had poor survival, had a presenting tumor that was ≥3.5 cm in greatest dimension (range 3.5–9.5 cm). The median survival in these four cases was 6 months regardless of therapy. One of these patients (Case 3) achieved temporary remission following three cycles of chemotherapy (CHOP). Within 5 months, however, the patient relapsed with aggressive disease, which was unresponsive to further treatment. Aside from disease stage and tumor size, there were no readily identifiable clinical features that distinguished patients with localized disease and good prognosis, and those with extensive disease and aggressive clinical course.
Histologic Findings
The cases showed similar histologic features (see Figures 1, 2, 3 and 4). The extranodal cases showed infiltrative margins with focal destruction of the surrounding normal tissues. The lymph node specimen similarly showed near complete replacement of the node with focal extracapsular extension. All of the tumors demonstrated a diffuse growth pattern composed of large polygonal cells. Although the cells were predominantly arranged in discohesive sheets, focal spindling was noted in three cases and was prominent in one (Case 4, see Figure 4). The neoplastic cells were large, with moderate to abundant eosinophilic cytoplasm, round to irregular nuclei with vesicular chromatin, and one to several distinct nucleoli. In addition, Cases 2 and 4 contained occasional cells with prominent nuclear grooves. Cellular atypia was mild to moderate with two cases showing pleomorphic and occasional anaplastic cells (Cases 3 and 4). Hemophagocytosis was identified within the tumor cells of three cases, however, this feature was only conspicuous in one (Case 1). Mitotic activity was highly variable throughout the five cases. Geographic necrosis was noted in the lymph node specimen (Case 5). Infiltrating neutrophils, lymphocytes, and occasional eosinophils were present and were focally abundant in the background in four of the cases.

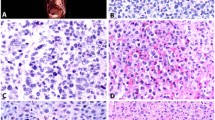
Case 1: histiocytic sarcoma of the palate with typical morphology. (a) Discohesive sheets of large, polygonal cells invading bone ( × 100); (b) Prominent hemophagocytosis within neoplastic cells (arrows) ( × 400).

Case 2: metastatic soft tissue mass of the forehead. (a) Histiocytic sarcoma diffusely infiltrating skeletal muscle (x100); (b) Many neoplastic cells with convoluted nuclear contours and occasional grooves, and virtual absence of a background of inflammatory cells (x400); (c) Representative immunohistochemical stains demonstrate reactivity for CD45, (d) CD163 (cytoplasmic and membranous, finely granular), (e) CD68, (f) Lysozyme (cytoplasmic, granular), and (g) MAC387; (h) No immunoreactivity for S-100 was identified ( × 400).

Case 3: sigmoid colon tumor with nuclear pleomorphism, mitotic activity and occasional foci of hemophagocytosis (arrows) ( × 100).

Case 4: histiocytic sarcoma of the colon with area of prominent spindling ( × 100).
Immunophenotypic Findings
The immunohistochemical features are listed in Table 3, and representative photomicrographs of Case 2 are depicted in Figures 2c–h. As one of the inclusion criteria, all specimens showed immunoreactivity for CD45, although expression was diffusely weak in two cases. Some variability in expression of histiocytic markers was noted; however, CD163 and CD68 consistently showed intense immunoreactivity. Lysozyme was weakly reactive in two cases, and MAC387 expression was somewhat inconsistent (2/5 positive, 1/5 focally positive, 2/5 negative). Scattered cells showed reactivity for factor XIIIa in two of the tumors. Whether these cells were a subset of the malignant population or were merely part of the non-neoplastic background was uncertain. S-100 was focally positive in four cases with one of these specimens showing more extensive areas of reactivity. All cases were negative for HMB-45, tyrosinase, and Melan A. Ki-67 immunoreactivity was variable, staining 1–15% of malignant nuclei. All remaining B- and T-cell, epithelial, melanocytic, and accessory/dendritic cell markers were uniformly negative.
Molecular Genetic Findings
Results of the IgH, TCRβ, and TCRγ gene rearrangement studies are summarized in Table 4 . Monoclonal IgH bands were identified in two cases and both IgH and TCRγ gene rearrangements were seen in one case. In all cases in which monoclonal bands were detected, B or T lymphocytes comprised <5%, and in most cases <1%, of the tissue cellularity. A small focus of residual lymph node was seen in Case 5. DNA was not amplifiable in Case 2, most likely due to low numbers of lymphocytes.
Electron Microscopic Findings
Ultrastructural analysis was carried out on three specimens (Cases 1, 3, and 5). The results in these cases were nonspecific and included scattered lysosomes within the cytoplasm, consistent with histiocytic origin. No Birbeck granules or desmosomes were observed. Occasional cytoplasmic processes were identified in Case 5 (inguinal lymph node); however, well-formed cellular junctions were not identified.
Discussion
HSs are a problematic and controversial group of rare hematopoietic neoplasms. Clinically, these tumors appear to show some distinctive features including frequent extranodal presentation, poor prognosis19, 20, 33, 41, 50, 51 and, as suggested by a recent study,52 in addition to the current one, a possible relationship between tumor size and poor prognosis. The evolution of the diagnosis of HS, as it is currently defined, is the product of continually improving diagnostic tools and more stringent diagnostic criteria. Despite its rarity, however, a growing number of reports have credibly documented the occurrence of these histiocytic tumors in nodal30, 31, 41 and a variety of extranodal sites including the gastrointestinal tract,35, 37, 39, 41, 42 skin,9, 33, 36, 38, 41 bone marrow,34 spleen,32 and central nervous system.40, 43, 44
In this study, we describe five neoplasms with the histopathologic features of true histiocytic origin. The diagnosis was based on morphology supported by an extensive immunophenotypic analysis that verified the expression in at least two histiocytic antigens and excluded other large cell malignancies in the differential diagnosis. Although light microscopy alone is unreliable in establishing the diagnosis, the cases in the current study and review of literature describe similar histologic features consisting of discohesive large cells with pleomorphic nuclei and abundant cytoplasm.2, 13, 17, 20, 28, 41, 50, 51, 52, 53 Malignant cytologic features with evidence of tissue invasion or destruction were constant findings in this study and are necessarily present in order to distinguish from benign histiocytic proliferations.18, 19, 20 Hemophagocytosis, a feature previously felt to be important in the diagnosis, may be present but is nonspecific29, 54, 55 and frequently is not prominent in bona fide cases.1, 17, 19, 20, 41, 51 Likewise, this feature was inconspicuous in all but one of our cases.
The importance of immunophenotypic studies in the diagnosis of histiocytic neoplasms has been emphasized.13, 17, 51 However, establishing histiocytic lineage by special stains has historically been elusive. Markers once thought to be specific for histiocytic differentiation, such as alpha-1-antitrypsin, alpha-1-antichymotrypsin, lysozyme, and KP1, have been shown to have low specificity and may be positive in a wide variety of neoplasms, including a significant percentage of non-Hodgkin lymphomas.6, 7, 20, 22, 23, 24, 56, 57 Moreover, CD68 (KP1) and alpha-1-antitrypsin immunoreactivity have been reported in melanoma with similar frequency to that of traditional melanoma markers (ie S-100, HMB-45), although the staining is usually not as intense.57, 58 Monoclonal antibodies, such as PG-M1,59 and more recently CD163,60, 61, 62 have demonstrated more restricted reactivity. In particular, CD163 has recently been studied in a variety of benign and malignant tissues and shows almost exclusive expression in cells of monocyte/macrophage lineage.26, 63 A single study of cultured uveal melanomas showed expression of both CD68 and CD163 in the cultured cells although the tissue from the uveal melanoma was not immunoreactive with CD163.64 In concordance with these studies, strong cytoplasmic immunoreactivity for CD163 was present in all of the cases in the current study, attesting to their histiocytic differentiation. This antigen has been identified as a hemoglobin scavenger receptor involved in the removal of free hemoglobin from the plasma as well as the uptake of iron in tissue macrophages.25, 62, 65 In addition to this role in iron homeostasis, CD163 is also believed to have an anti-inflammatory function since it is rapidly upregulated in the presence of IL-10 and the glucocorticoid dexamethasone.25, 61, 62, 65, 66, 67
In addition to establishing histiocytic origin, immunohistochemistry plays an equally important and challenging exclusionary role in evaluating the differential diagnosis of histiocytic sarcoma. Our immunohistochemical analysis included an extensive panel of several epithelial, lymphoma, melanoma, and dendritic cell antibodies. Certain lymphoid markers, such as CD4 and CD22, were excluded since expression of these T- and B-cell markers may be expressed in some cells of myelomonocytic or histiocytic lineage.68, 69, 70 S-100, on the other hand, was a necessary component of the immunohistochemical analysis but may demonstrate variable results in histiocytic tumors. Immunoreactivity for this antibody in our study, therefore, required additional immunohistochemical stains to confidently exclude melanoma (ie melan A, HMB-45, tyrosinase), carcinoma (ie cytokeratin), and accessory/dendritic cell tumors (ie CD1a, CD21, CD35).2, 51, 71
S-100 reactivity has been reported in benign and malignant histiocytic proliferations,19, 41, 43, 51 although usually the degree of reactivity, as in the present study, is less than that seen in other accessory/dendritic cell tumors (ie Langerhans cell histiocytosis, interdigitating dendritic cell tumor).51 Immunohistochemistry and, if necessary, electron microscopy can usually delineate these lesions. However, antigen expression of some histiocytic and dendritic cell subsets appear similar using monoclonal antibodies, likely reflecting their close functional relationship.72 This phenomenon was observed in Case 5, which showed histologic, immunophenotypic, and ultrastructural characteristics of both HS and interdigitating dendritic cell sarcoma, though the preponderance of features favored the former. This specimen also showed the greatest degree of S-100 immunoreactivity as compared to the focal or absent staining noted in the other cases. As previously observed,37, 41 this phenomenon may indicate that some histiocytic neoplasms show differentiation toward antigen-presenting cells (ie interdigitating cells) or that these tumors may arise from a common cell of origin.
An interesting and novel finding in this study was the presence of focal factor XIIIa immunoreactivity. Although the identity of the positive cells as neoplastic or benign was difficult to determine, the expression of factor XIIIa in a subset of the histiocytes is not entirely unexpected as factor XIIIa expression has also been reported in Erdheim–Chester disease, a rare non-Langerhans’ cell histiocytosis.73 The significance of this finding and its possible role in the histogenesis of HSs is uncertain and requires investigation in a greater number of cases.
Since the mature tissue histiocyte is believed to be the benign counterpart of HS, CD45 (LCA) immunoreactivity has been recommended and was used in this study as a requirement to establish hematopoietic origin. It also allowed differentiation from morphologically similar soft-tissue neoplasms.29 As not all histiocytes necessarily stain with CD45, however, the relevance of this criterion has been debated.74 While CD45 immunoreactivity may not be an absolute requirement with the aid of additional supportive studies such as electron microscopy,42 differentiation from malignant fibrous histiocytoma (undifferentiated sarcoma) may be extremely difficult. These tumors are believed to arise from undifferentiated mesenchymal cells rather than histiocytes, and typically show pleomorphic and storiform growth patterns.75, 76 Enzymatic and immunophenotypic studies77, 78 have shown that the neoplastic cells in these tumors are negative for histiocytic markers but may contain numerous reactive histiocytes. Despite these subtle morphologic and immunophenotypic differences, clinical history may be the only means of differentiating HS from a metastatic soft-tissue malignancy involving lymph node.19 Although clinical history was somewhat limited in this study, the case involving lymph node showed immunophenotypic and ultrastructural features most compatible with a primary histiocytic tumor.
Another unresolved topic regarding the diagnosis of HS is the necessity and utility of molecular gene rearrangement studies. The difficulties are twofold: (1) there are no universal molecular genetic markers for detection of clonal histiocytic proliferations, and (2) the interpretation of IgH and TCR clonality studies can be problematic. While one would expect the histiocytic proliferations to lack clonal IgH or TCR rearrangements, in fact, these have been commonly reported in HSs27, 28 and were also seen in this study. Monoclonal gene rearrangements were detected in three of five cases in this study, the remaining cases being not amplifiable for IgH. This phenomenon may represent a relatively common feature of malignant histiocytes, either lineage promiscuity related to the primitive nature of these cells79, 80 or dual genotype in which some histiocytes may abnormally retain the ability to rearrange the TCR and/or IgH genes despite phenotypically developing along histiocytic lines.28 The former explanation is supported by the observation that histiocytic neoplasms occasionally arise in association with mediastinal germ-cell tumors81, 82 and that IgH rearrangements have been previously reported in myeloid sarcoma.83, 84
Alternatively, cross-contamination from positive cases or ‘pseudomonoclonality’ due to extremely low numbers of lymphocytes may contribute to false positive molecular testing results.85, 86 Although lymphocytes were sparse in the specimens in the present study, duplicate PCR testing decreases the likelihood of this pitfall. Finally, the presence of small clonal-like lymphoid populations, perhaps due to an immunologic response to the tumor,87 is another consideration that may result in clonal gene rearrangements studies.
The possibility of the gene rearrangement studies defining tumor lineage as well as clonality, was also considered. However, classifying the tumors in our study as poorly differentiated lymphomas would imply a complete loss of all B- and T-cell antigens tested in our immunohistochemical panel, in addition to aberrant and exclusive expression of multiple histiocyte markers to include CD163. Therefore, we favor these neoplasms being of histiocytic origin based on the overwhelming phenotypic data and believe the results of the gene rearrangement studies most likely reflect an inherent property of the malignant histiocytes. Further studies involving microdissection of individual tumor cells followed by PCR amplification and sequence analysis may provide a more definitive explanation of these results.
Although the number of cases in this study is small, several conclusions may be drawn, which contribute to and further refine the clinicopathologic features of HS. The presentation of these rare hematopoietic tumors is more commonly extranodal, particularly involving the gastrointestinal tract. The clinical course is frequently aggressive with an associated high mortality rate. Stage of disease and possibly tumor size appear to be prognostically significant. Overlapping histologic, immunophenotypic, and ultrastructural features between HS and the dendritic cell neoplasms suggest a close relationship in the histogenesis of these tumors. Gene rearrangement studies remain a controversial issue but are probably not necessary to establish the diagnosis. Results must be interpreted in light of the clinical, morphologic, and immunophenotypic features. Continued investigation is necessary to fully understand the nature of these genetic findings and to further elucidate the pathogenesis of these neoplasms. Finally, the morphologic and immunophenotypic features are relatively uniform, however, an extensive panel of immunohistochemical markers is necessary to adequately exclude more common, poorly differentiated malignancies. Until the recent characterization of CD163, histiocytic markers have typically demonstrated poor specificity and have confused the diagnosis of histiocytic neoplasms. CD163 is a sensitive and apparently restricted marker, with a rare exception, for identifying lesions of histiocytic lineage and is, therefore, important in the diagnosis of HS.
Disclaimer
The opinions and assertions contained herein are the private views of the authors and are not to be considered as official or as representing the views of the Departments of the Army or Defense.
References
Jaffe ES . Histiocytoses of lymph nodes: biology and differential diagnosis. Semin Diagn Pathol 1988;5:376–390.
Jaffe ES, Harris NL, Stein H, Vardiman JW (eds). Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARCPress: Lyon 2001. pp 273–290.
Gall EA, Mallory TB . Malignant lymphoma. A clinico-pathologic survey of 618 cases. Am J Pathol 1942;18:381–429.
Rappaport H . Tumors of the hematopoietic system. Section 3 3, fascicle 8. In: Atlas of Tumor Pathology. US Armed Forces Institute of Pathology: Washington, DC, 1966, 442pp.
Morris MW, Davey FR . Immunologic and cytochemical properties of histiocytic and mixed histiocytic–lymphocytic lymphomas. Am J Clin Pathol 1975;63:403–414.
Isaacson PG, Spencer J, Connolly CE, et al. Malignant histiocytosis of the intestine: A T-cell lymphoma. Lancet 1985;2:688–691.
Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin's disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed–Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985;66:848–858.
van der Valk P, van Oostveen JW, Stel HV, et al. Phenotypic and genotypic analysis of large-cell lymphomas, formerly classified as true histiocytic lymphoma: identification of an unusual group of tumors. Leuk Res 1990;14:337–346.
Aria E, Su WPD, Roche PC, et al. Cutaneous histiocytic malignancy. Immunohistochemical re-examination of cases previously diagnosed as cutaneous ‘histiocytic lymphoma’ and ‘malignant histiocytosis’. J Cutan Pathol 1993;20:115–120.
Egeler RM, Schmitz L, Sonneveld P, et al. Malignant histiocytosis: a reassessment of cases formerly classified as histiocytic neoplasms and review of the literature. Med Pediatr Oncol 1995;25:1–7.
Mathé G, Gerard-Marchant R, Texier JL, et al. The two varieties of lymphoid tissue ‘reticulosarcomas’, histiocytic and histioblastic types. Br J Cancer 1970;24:687–695.
Jaffe ES, Braylan RC, Nanba K, et al. Functional markers: a new perspective on malignant lymphomas. Cancer Treat Rep 1977;61:953–962.
Turner RR, Wood GS, Beckstead JH, et al. Histiocytic malignancies: morphologic, immunologic, and enzymatic heterogeneity. Am J Surg Pathol 1984;8:485–500.
Kamel OW, Gocke CD, Kell DL, et al. True histiocytic lymphoma: a study of 12 cases based on current definition. Leuk Lymphoma 1995;18:81–86.
Picker LJ, Weiss LM, Medeiros LJ, et al. Immunophenotypic criteria for the diagnosis of non-Hodgkin's lymphoma. Am J Pathol 1987;128:181–201.
Weiss LM, Picker LJ, Copenhaver CM, et al. Large-cell hematolymphoid neoplasms of uncertain lineage. Hum Pathol 1988;19:967–973.
Lauritzen AF, Delsol G, Hansen NE, et al. Histiocytic sarcomas and monoblastic leukemias: a clinical, histologic, and immunophenotypical study. Am J Clin Pathol 1994;102:45–54.
Risdall RJ, McKenna RW, Nesbit ME, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 1979;44:993–1002.
Gonzalez CL, Jaffe ES . The histiocytoses: clinical presentation and differential diagnosis. Oncology 1990;4:47–60.
Jaffe ES . Malignant histocytosis and true histiocytic lymphoma. In: Jaffe ES (ed). Surgical Pathology of Lymph and Related Organs. Philadelphia: Saunders, 1995, pp 560–593.
Cattoretti G, Villa A, Vezzoni P, et al. Malignant histiocytosis: a phenotypic and genotypic investigation. Am J Pathol 1990;136:1009–1019.
Soini Y, Miettinen M . Widespread immunoreativity for alpha-1-antichymotrypsin in different types of tumors. Am J Clin Pathol 1988;90:131–136.
Banks PM, Metter J, Allred DC . Anaplastic large cell (Ki-1) lymphoma with histiocytic phenotype simulating carcinoma. Am J Clin Pathol 1990;94:445–452.
Carbone A, Gloghini A, Volpe R, et al. KP1 (CD68)-positive large cell lymphomas: a histopathologic and immunophenotypic characterization of 12 cases. Hum Pathol 1993;24:886–896.
Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature 2001;409:198–201.
Walter RB, Bächli EB, Schaer DJ, et al. Expression of the hemoglobin scavenger receptor (CD163/HbSR) as immunophenotypic marker of monocytic lineage in acute myeloid leukemia. Blood 2003;101:3755–3756.
Weiss LM, Trela M, Cleary ML, et al. Frequent immunoglobulin and T-cell receptor gene rearrangements in ‘histiocytic’ neoplasms. Am J Pathol 1985;121:369–373.
Hanson CA, Jaszcz W, Kersey JH, et al. True histiocytic lymphoma: histopathologic, immunophenotypic and genotypic analysis. Br J Haematol 1989;73:187–198.
Levine EG, Hanson CA, Jaszcz W, et al. True histiocytic lymphoma. Semin Oncol 1991;18:39–49.
Mirchandani I, Shah I, Palutke M, et al. True histiocytic lymphoma: a report of four cases. Cancer 1983;52:1911–1918.
Thomas P, Said JW, Rosenfelt FP, et al. True histiocytic lymphoma: an immunohistochemical and ultrastructural study of two cases. Am J Clin Pathol 1984;81:243–248.
Franchino C, Reich C, Distenfeld A, et al. A clinicopathologically distinctive primary splenic histiocytic neoplasm. Demonstration of its histiocytic derivation by immunophenotypic and molecular genetic analysis. Am J Surg Pathol 1988;12:398–404.
Ralfkiaer E, Delsol G, O’Connor NTJ, et al. Malignant lymphomas of true histiocytic origin. A clinical, histological, immunophenotypic and genotypic study. J Pathol 1990;160:9–17.
Salyer J, Craven CM . Malignant histiocytosis in a patient with acquired immunodeficiency syndrome-related complex. Arch Pathol Lab Med 1990;114:376–378.
Milchgrub S, Kamel OW, Wiley E, et al. Malignant histiocytic neoplasms of the small intestine. Am J Surg Pathol 1992;16:11–20.
Soria C, Orradre JL, García-Almagro D, et al. True histiocytic lymphoma (monocytic sarcoma). Am J Dermatopathol 1992;14:511–517.
Miettinen M, Fletcher CDM, Lasota J . True histiocytic lymphoma of small intestine: an analysis of two S-100 protein-positive cases with features of interdigitating reticulum cell sarcoma. Am J Clin Pathol 1993;100:285–292.
Osborne BM, MacKay B . True histiocytic lymphoma with multiple skin nodules. Ultrastruct Pathol 1994;18:241–246.
Álvaro T, Bosch R, Salvadó MT, et al. True histiocytic lymphoma of the stomach associated with low-grade B-cell mucosa-associated lymphoid tissue (Malt)-type lymphoma. Am J Surg Pathol 1996;20:1406–1411.
Torres CF, Korones DN, Powers JM, et al. Primary leptomeningeal histiocytic lymphoma in a young child. Med Pediatr Oncol 1996;27:547–550.
Copie-Bergman C, Wotherspoon AC, Norton AJ, et al. True histiocytic lymphoma. A morphologic, immunohistochemical, and molecular genetic study of 13 cases. Am J Surg Pathol 1998;22:1386–1392.
Seo IS, Henley JD, Min KW, et al. True histiocytic lymphoma of the esophagus in an HIV-positive patient: an ultrastructural study. Ultrastruct Pathol 1999;23:333–339.
Cheuk W, Walford N, Lou J, et al. Primary histiocytic lymphoma of the central nervous system. A neoplasm frequently overshadowed by a prominent inflammatory component. Am J Surg Pathol 2001;25:1372–1379.
Sun W, Nordberg ML, Fowler MR . Histiocytic sarcoma involving the central nervous system: clinical, immunohistochemical, and molecular genetic studies of a case with review of the literature. Am J Surg Pathol 2003;27:258–265.
Trainor KJ, Brisco MJ, Wan JH, et al. Gene rearrangement in B- and T-lymphoproliferative disease detected by the polymerase chain reaction. Blood 1991;78:192–196.
Reed TJ, Reid A, Wallberg K, et al. Determination of B-cell clonality in paraffin-embedded lymph nodes using the polymerase chain reaction. Diagn Mol Pathol 1993;2:42–49.
Abruzzo LV, Griffith LM, Nandedkar M, et al. Histologically discordant lymphomas with B-cell and T-cell components. Am J Clin Pathol 1997;108:316–323.
Krafft AE, Taubenberger JK, Sheng ZM, et al. Enhanced sensitivity with a novel TCRγ PCR assay for clonality studies in 569 formalin-fixed, paraffin-embedded (FFPE) cases. Mol Diagn 1999;4:119–133.
Dykstra MJ (ed). Biological Electron Microscopy. Theory, Techniques, and Troubleshooting. Plenum Press: New York, 1992, 359pp.
van der Valk, te Velde J, Jansen J, et al. Malignant lymphoma of true histiocytic origin: histiocytic sarcoma. A morphological, ultrastructural, immunological, cytochemical and clinical study of 10 cases. Virchows Arch A Pathol Anat Histol 1981;391:249–265.
Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002;41:1–29.
Hornick JL, Jaffe ES, Fletcher CDM . Histiocytic sarcoma: a study of 13 extranodal cases [abstract]. Modern Pathol 2004;1 (Suppl 1):16A.
Isaacson P, Wright DH, Jones DB . Malignant lymphoma of true histiocytic (monocyte/macrophage) origin. Cancer 1983;51:80–91.
Kadin ME, Kamoun M, Lamberg J . Erythrophagocytic Tγ lymphoma: a clinicopathologic entity resembling malignant histiocytosis. N Engl J Med 1981;304:648–653.
Jaffe ES, Costa J, Fauci AS, et al. Malignant lymphoma and erythrophagocytosis simulating malignant histiocytosis. Am J Med 1983;75:741–749.
Soini Y, Miettinen M . Alpha-1-antitrypsin and lysozyme: their limited significance in fibrohistiocytic tumors. Am J Clin Pathol 1989;91:515–521.
McHugh M, Miettinen M . KP1 (CD68). Its limited specificity for histiocytic tumors. Appl Immunohistochem 1994;2:186–190.
Pernick NL, DaSilva M, Gangi MD, et al. Histiocytic markers in melanoma. Mod Pathol 1999;12:1072–1077.
Falini B, Flenghi L, Pileri S, et al. PG-M1: a new monoclonal antibody directed against a fixative-resistant epitope on the macrophage-restricted form of the CD68 molecule. Am J Pathol 1993;142:1359–1372.
Droste A, Sorg C, Högger P . Shedding of CD163, a novel regulatory mechanism for a member of the scavenger receptor cysteine-rich family. Biochem Biophys Res Commun 1999;256:110–113.
Ritter M, Buechler C, Langmann T, et al. The scavenger receptor CD163: regulation, promoter structure and genomic organization. Pathobiology 1999;67:257–261.
Graversen JH, Madsen M, Moestrup SK . CD163: a signal receptor scavenging haptoglobin-hemoglobin complexes from plasma. Int J Biochem Cell Biol 2002;34:309–314.
Nguyen TT, Schwartz EJ, West RB, et al. Expression of CD163 in normal tissues, lymphomas, carcinomas and sarcomas is limited to a few select neoplasms [abstract]. Mod Pathol 2004;17 (Suppl 1):263A.
Woodward J, Reeves SK, Nichols C, et al. Evidence of macrophage and lymphocyte, but not dendritic cell, infiltration in posterior uveal melanomas, whilst cultured uveal melanomas demonstrate pluripotency by expressing CD68 and CD163. In J Exp Pathol 2004;85:35–43.
Sulahian TH, Högger P, Wahner AE, et al. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine 2000;12:1312–1321.
Högger P, Dreier J, Droste A, et al. Identification of the integral membrane protein RM3/1 on human monocytes as a glucocorticoid-inducible member of the scavenger receptor cysteine-rich family (CD163). J Immunol 1998;161:1883–1890.
Schaer DJ, Boretti FS, Hongegger A, et al. Molecular cloning and characterization of the mouse CD163 homologue, a highly glucocorticoid-inducible member of the scavenger receptor cysteine-rich family. Immunogenetics 2001;53:170–177.
Wood GS, Warner NL, Warnke RA . Anti-Leu-3/T4 antibodies react with cells of monocyte/macrophage and Langerhans lineage. J Immunol 1983;131:212–216.
Hsu S-M, Hsu P-L . Aberrant expression of T cell and B cell markers in myelocyte/monocyte/histiocyte-derived lymphoma and leukemia cells. Am J Pathol 1989;134:203–212.
Escribano L, Orfao A, Villarrubia J, et al. Expression of lymphoid-associated antigens in mast cells: report of a case of systemic mast cell disease. Br J Haematol 1995;91:941–943.
Andriko JW, Kaldjian EP, Tsokos M, et al. Reticulum cell neoplasms of lymph nodes. A clinicopathologic study of 11 cases with recognition of a new subtype derived from fibroblastic reticular cells. Am J Surg Pathol 1998;22:1048–1058.
Franklin WA, Mason DY, Pulford K, et al. Immunohistological analysis of human mononuclear phagocytes and dendritic cells by using monoclonal antibodies. Lab Invest 1986;54:322–335.
Rush WL, Andriko JW, Galateau-Salle F, et al. Pulmonary pathology of Erdheim–Chester Disease. Mod Pathol 2000;13:747–754.
Gatter KC, Abdulaziz Z, Beverley P, et al. Use of monoclonal antibodies for the histopathological diagnosis of human malignancy. J Clin Pathol 1982;35:1253–1267.
Weiss SW, Enzinger FM . Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer 1978;41:2250–2266.
Roholl PJM, Kleyne J, Elbers H, et al. Characterization of tumour cells in malignant fibrous histiocytomas and other soft tissue tumours in comparison with malignant histiocytes. I. Immunohistochemical study on paraffin sections. J Pathol 1985;147:87–95.
Wood GS, Beckstead JH, Turner RR, et al. Malignant fibrous histiocytoma tumor cells resemble fibroblasts. Am J Surg Pathol 1986;10:323–335.
Soini Y, Miettinen M . Immunohistochemistry of markers of histiomonocytic cells in malignant fibrous histiocytomas: a monoclonal antibody study. Pathol Res Pract 1990;186:759–767.
Greaves MF, Chan LC, Furley AJW, et al. Lineage promiscuity in hemopoietic differentiation and leukemia. Blood 1986;67:1–11.
Cumano A, Paige CJ, Iscove NN, et al. Bipotential precursors of B cells and macrophages in murine fetal liver. Nature 1992;356:612–615.
deMent SH . Association between mediastinal germ cell tumors and hematologic malignancies: an update. Hum Pathol 1990;21:699–703.
Margolin K, Traweek T . The unique association of malignant histocytosis and a primary gonadal germ cell tumor. Med Pediatr Oncol 1992;20:162–164.
Kyoda K, Nakamura S, Matano S, et al. Prognostic significance of immunoglobulin heavy chain gene rearrangement in patients with acute myelogenous leukemia. Leukemia 1997;11:803–806.
Schmetzer HM, Braun S, Wiesner D, et al. Gene rearrangements in bone marrow cells of patients with acute myelogenous leukemia. Acta Hematol 2000;103:125–134.
Elenitoba-Johnson KSJ, Bohling SD, Mitchell RS, et al. PCR analysis of the immunoglobulin heavy chain gene in polyclonal processes can yield pseudoclonal bands as an artifact of low B cell number. J Mol Diagn 2000;2:92–96.
Bagg A . Clinical applications of molecular genetic testing in hematologic malignancies: advantages and limitations. Hum Pathol 2003;34:352–358.
Lim SH, Badros A, Lue C, et al. Distinct T-cell clonal expansion in the vicinity of tumor cells in plasmacytoma. Cancer 2001;91:900–908.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented in part at the 93rd annual meeting of the United States and Canadian Academy of Pathology, Vancouver, British Columbia, March 6–12, 2004.
Rights and permissions
About this article
Cite this article
Vos, J., Abbondanzo, S., Barekman, C. et al. Histiocytic sarcoma: a study of five cases including the histiocyte marker CD163. Mod Pathol 18, 693–704 (2005). https://doi.org/10.1038/modpathol.3800346
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800346
Keywords
This article is cited by
-
TYMS promotes genomic instability and tumor progression in Ink4a/Arf null background
Oncogene (2023)
-
Lymph node recurrence and re-excision after primary tumor resection of a histiocytic sarcoma of duodenal origin: a case report
Surgical Case Reports (2022)
-
Primary splenic histiocytic sarcoma successfully treated with splenectomy: a case report and literature review
International Cancer Conference Journal (2022)
-
A case of recurrent refractory cervical primary histiocytic sarcoma treated with pembrolizumab
International Cancer Conference Journal (2022)
-
Nodal histiocytic sarcoma with prominent eosinophilic infiltration: expression of eotaxin-2 on tumor cells
Diagnostic Pathology (2021)
