
Abstract
Spinocerebellar ataxia (SCA) is a group of dominantly inherited heterogeneous disorders in which 43 subtypes have been identified to date. Recently, Japanese and French families with SCA type 42 (SCA42) were found to have a missense mutation (c.5144G>A; R1715H) in CACNA1G. We performed genetic analysis of 84 unrelated families to find the prevalence of SCA42 in Japan. Two families were found to have the previously reported missense mutation. Clinical presentations of the affected members of these families were similar to those of the previously reported French and Japanese families. Our study demonstrates that SCA42 exists in small numbers in Japan, and further supports the idea that SCA42 is a slowly progressive, pure cerebellar ataxia.
Similar content being viewed by others


Introduction
Spinocerebellar ataxia (SCA) is a genetically inherited heterogeneous neurodegenerative disorder that affects the cerebellum, brainstem, spinal cord and cranial nerve nuclei, resulting in progressive cerebellar ataxia of the gait and limbs. It is variably associated with nystagmus, dysarthria, intention tremor and ophthalmoparesis. Disease onset is usually between 30 and 50 years of age, although early onset in childhood and onset after 60 years have been reported. The clinical diagnosis of SCA subtypes is complicated by their overlapping of their phenotypic variation. To date, 43 different genetic loci for SCA have been reported and a causative gene has been identified for most. In 2015, SCA type 42 (SCA42) was reported to be caused by a voltage-gated calcium channel 1G (CACNA1G) mutation. Japanese1 and French2 families in two separate studies were found to have a heterozygous missense mutation in CACNA1G (c.5144G>A; R1715H).
In the present study, we examined the prevalence and incidence of SCA42 in Japan. We performed genetic analyses on individuals whose SCA subtype was unknown.
Subjects and methods
Genetic analyses were performed on 84 unrelated Japanese families with SCA, who presented to our hospital between April 1999 and September 2016. Previously, we published a similar study on patients who presented until March 1999.3 In all cases, progressive cerebellar ataxia was the cardinal clinical manifestation. The majority of patients presented with an adult onset phenotype. Brain magnetic resonance imaging (MRI) showed cerebellar atrophy or pontocerebellar atrophy. All procedures were approved by the Hokkaido University Ethics Committee, and informed consent was obtained from all participants (healthy/asymptomatic and diseased).
Genomic DNA was extracted from blood lymphocytes by standard methods. Abnormal expansions of tri-, penta- and hexa-nucleotide repeats or reported mutations in SCA1, SCA2, Machado–Joseph disease/SCA3, SCA6, SCA7, SCA8, dentatorubal-pallidoluysion atrophy, SCA14, SCA17, SCA31 and SCA36 genes were excluded in all 84 probands. Direct sequencing of all exons of CACNA1G for SCA42 was performed (OMIM #616795). After treatment with shrimp alkaline phosphatase and exonuclease I, polymerase chain reaction samples were sequenced directly (BigDye Terminator, version 3.1, and 3130 GeneticAnalyzer; Applied Biosystems, Tokyo, Japan).
Results
Eighty-four SCA families were examined for CACNA1G mutations in all 38 exons, and two families were positive. Both families had the previously reported pathogenic missense mutation (c.5144 G>A; R1715H),1, 2 which was also confirmed by restriction fragment length polymorphism analysis (Supplementary data). No other mutations were found. Family 1 had previously been reported as having a novel SCA with a possible linkage to the SCA12 locus (5q32).4 Affected members of this family presented with a pure cerebellar phenotype.4 In our current analysis, all affected members and one asymptomatic carrier (at the time of presentation; previously reported as II-6) were found to have the pathogenic mutation.
The pedigree of the other family, family 2, is shown (Figure 1). Two members (IV-6 and IV-8) still visit a nearby hospital, as their mother (III-6) had previously. All three developed gait disturbance, mild-to-moderate speech difficulties, and smooth pursuit defects in their mid-30s. IV-6 and III-6 eventually became wheelchair bound. They also had the pathogenic mutation. Their clinical features are shown in Table 1. Neuroradiological examinations revealed atrophy confined only to the cerebellum in all three individuals (Figure 2); therefore, they were also diagnosed with pure cerebellar ataxia.

Pedigree of family 2. Males are represented as squares. Females are represented as circles. Black-filled circles represent affected individuals, while open circles indicate unaffected members. Genetic analysis was performed in the proband (III-6), and her two daughters (IV-6, IV-8).

Neuroradiological findings (a): Mid-sagittal (T1WI) MRI of the brain of patient III-6 from family 2. (b): Mid-axial CT scan of the brain of patient IV-6 from family 2. (c): Mid-sagittal (T1WI) MRI of the brain of patient IV-8 from family 2.
Discussion
We identified two Japanese families with a previously reported pathologic missense mutation. This is in addition to the Japanese family reported by Morino et al.,1 indicating that SCA42 is present in small numbers in Japan. In contrast to Morino, who examined families in Hiroshima (southwest Japan), our patients were from Hokkaido (northernmost prefecture of Japan). Analyses of other regions in Japan are warranted to determine the prevalence of this particular subtype and its impact.
To date, this disease has only been reported in France and Japan, and this report would be the third. It is unknown why it is specific to these countries, and it is possible that SCA42 is present in other locations, but has not been reported. Further epidemiological studies are warranted to better understand this rare subtype.
Our study supports the clinical presentations seen in the previously reported SCA42 families: slowly progressive, pure cerebellar ataxia accompanied with dysarthria, gait disturbance and abnormal eye movements. Many patients with SCA subtypes tend to present with nystagmus, especially those with pure cerebellar ataxia. SCA6 patients present with a variety of nystagmus types, while downbeat positioning nystagmus is rare in SCA31 patients.5 Nystagmus did not present in our patients, although saccadic pursuits were common.
Age of onset varied from 9–78 years in Coutelier’s patients and 20–70 years in Morino’s patients.1, 2 In contrast, the age of onset in our patients was 13–38 years (average, 31.8 years), which may provide clues to the unknown current status of the asymptomatic carrier (II-6) of family 1,4 who was 34 years of age at presentation and found to have this mutation in SCA42.
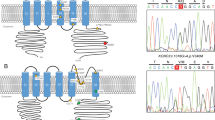
The c.5144G>A mutation is located in the S4 segment, which is the voltage sensor of Cav3.1.1 Electrophysiological studies showed that activation of mutant Cav3.1 transfected into HEK293T cells shifted toward more positive membrane potentials, while the inactivation curves of Ca2+ were also shifted positively.1, 2 This mutation leads to a change in calcium channels, but specific details remain unknown. Further studies should assess why this mutation leads to this specific subtype.
The clinical presentations of SCA subtypes are similar, making a succinct diagnosis difficult. Genetic testing has proven to be invaluable in such patients. In family 1, previous genome-wide analysis suggested linkage to the SCA12 locus. However, subtype-specific genetic analysis identified a mutation at a different locus in the asymptomatic carrier.
In summary, we identified two additional Japanese families with SCA42. The combination of clinical assessments and genetic analyses is ideal for making a subtype diagnosis. Since there is no cure for SCA, identification of the causative genes for the remaining disease loci is important for designing treatment options to improve the function and quality of life of these patients.
References
Morino, H., Matsuda, Y., Muguruma, K., Miyamoto, R., Ohsawa, R., Ohtake, T. et al. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol. Brain 8, 89 (2015).
Coutelier, M., Blesneac, J., Monteil, A., Monin, M., Ando, K., Mundwiller, E. et al. A recurrent mutation in CACNA1G alters Cav3.1T-type calcium channel conduction and causes autosomal-dominant cerebellar ataxia. Am. J. Hum. Genet. 97, 726–737 (2015).
Basri, R., Yabe, I., Soma, H. & Sasaki, H. Spectrum and prevalence of autosomal dominant spinocerebellar ataxia in Hokkaido, the northern island of Japan: a study of 113 Japanese families. J. Hum. Genet. 52, 848–855 (2007).
Sato, K., Yabe, I., Fukuda, Y., Soma, H., Nakahara, Y., Tsuji, S. et al. Mapping of autosomal dominant cerebellar ataxia without the pathogenic PPP2R2B mutation to the locus for spinocerebellar ataxia 12. Arch. Neurol. 67, 1257–1262 (2010).
Yabe, I., Matsushima, M., Yoshida, K., Ichikawa, K., Shirai, S., Takahashi, I. et al. Rare frequency of downbeat positioning nystagmus in spinocerebellar ataxia type 31. J. Neurol. Sci. 350, 90–92 (2015).
Acknowledgements
We are very grateful to all the patients and their families for their willingness to participate in this study. This work was partly supported by a Grant-in-Aid for ‘The Research Committee for Ataxic Diseases’ of the Research on Measures for Intractable Diseases from the Ministry of Health, Welfare and Labour, Japan, and by a Grant-in- Aid from the Takeda Science Foundation.Takeda Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Kimura, M., Yabe, I., Hama, Y. et al. SCA42 mutation analysis in a case series of Japanese patients with spinocerebellar ataxia. J Hum Genet 62, 857–859 (2017). https://doi.org/10.1038/jhg.2017.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.51
This article is cited by
-
A Review of Ocular Movement Abnormalities in Hereditary Cerebellar Ataxias
The Cerebellum (2023)
-
Zonisamide can ameliorate the voltage-dependence alteration of the T-type calcium channel CaV3.1 caused by a mutation responsible for spinocerebellar ataxia
Molecular Brain (2020)
-
Genetic screening for potassium channel mutations in Japanese autosomal dominant spinocerebellar ataxia
Journal of Human Genetics (2020)
-
Neuronal Cav3 channelopathies: recent progress and perspectives
Pflügers Archiv - European Journal of Physiology (2020)
