
Abstract
Osteogenesis imperfecta (OI) is a group of clinically and genetically heterogeneous disorders characterized by decreased bone mass and recurrent bone fractures. Transmembrane protein 38B (TMEM38B) gene encodes trimeric intracellular cation channel type B (TRIC-B), mutations of which will lead to the rare form of autosomal recessive OI. Here we detected pathogenic gene mutations in TMEM38B and investigated its phenotypes in three children with OI from two non-consanguineous families of Chinese Han origin. The patients suffered from recurrent fractures, low bone mass, mild bone deformities and growth retardation, but did not have impaired hearing or dentinogenesis imperfecta. Next-generation sequencing and Sanger sequencing revealed a homozygous novel acceptor splice site variant (c.455-7T>G in intron 3, p.R151_G152insVL) in family 1 and a homozygous novel nonsense variant (c.507G>A in exon 4, p.W169X) in family 2. The parents of the probands were all heterozygous carriers of these mutations. We reported the phenotype and novel mutations in TMEM38B of OI for the first time in Chinese population. Our findings of the novel mutations in TMEM38B expand the pathogenic spectrum of OI and strengthen the role of TRIC-B in the pathogenesis of OI.
Similar content being viewed by others
Introduction
Osteogenesis imperfecta (OI) is a group of clinically and genetically heterogeneous disorders characterized by decreased bone mass, recurrent bone fractures, progressive bone deformity and growth retardation, which will significantly impair the activity abilities and quality of life of the patients.1 Extra-skeletal features of OI include hearing loss, blue sclera, dentinogenesis imperfecta and joint hyperlaxity.2 The phenotypic spectrum of OI ranges widely from mild to lethal. The majority of OI cases follow an autosomal dominant pattern of inheritance, and are caused by mutations in genes encoding the type I procollagen chains, COL1A1 (MIM120150) or COL1A2 (MIM 120160).1 With the assistance of molecular chaperones and endoplasmic reticulum (ER) enzymes, two procollagen α1 proteins and one procollagen α2 protein form a triple helix in the ER. Collagen type I is formed in the extracellular matrix from procollagen type I after removal of its carboxyl and amino terminals.3 Mutations of genes, which lead to defects in collagen processing, post-translational modification, folding and crosslinking, ossification and mineralization and osteoblast development, including IFITM5, TMEM38B, CRTAP, SERPINH, PPIB, P3H1, P4HB, SP7, SERPINF, FKBP10, BMP1, PLOD2, WNT1, SEC24D, PLS3, SPARC and CREB3L1,3, 4, 5, 6, 7, 8, 9 are all causes of OI. As far as we know, mutations of 19 pathogenic genes have been identified to lead to OI (http://www.le.ac.uk/ge/collagen/).
The mutation in transmembrane protein 38B (TMEM38B) gene was first reported to lead to autosomal recessive OI in 2012, which was known as progressively deforming type of OI according to Nosology and classification of genetic skeletal disorders.10, 11, 12, 13 Located on chromosome 9q31.2, TMEM38B encodes trimeric intracellular cation channel type B (TRIC-B), a protein with 291 amino-acid residues that is expressed in the ER of most mammalian tissues.14 TRIC-B possesses three transmembrane segments (TM) cross the whole lipid bilayer, and forms a trimeric ER-membrane cation channel (a K+ channel) synchronized with inositol trisphosphate-mediated release of calcium.15 TM1, TM2 and TM3 are thought to contribute to ion conduction, channel gating and correct protein folding, respectively.16
Only two mutations in TMEM38B have been reported in the 22 individuals with OI worldwide.11, 12, 13 A c.455_542del mutation in TMEM38B (p.Gly152Alafs∗5) was reported as the cause of autosomal recessive OI in three consanguineous Saudi Arabian families and three consanguineous southern Israeli families in 2012 and 2013, respectively.11, 12 Another pathogenic mutation, a homozygous 35-kb deletion involving exons 1 and 2 of TMEM38B, was confirmed in an 11-year-old girl with OI in 2014.13 So far, no information about TMEM38B mutation has been reported in Chinese patients with OI.
Bisphosphonates (BPs), synthetic analogs of inorganic pyrophosphate, are widely used to treat bone fragility in children with moderate to severe forms of OI. Several studies demonstrate that BP treatment can produce a suppressed osteoclast-mediated bone resorption, marked increase in areal bone mineral density (BMD) and radiologically apparent reshaping of compressed vertebral bodies.17 The efficacy of BPs on patients of TMEM38B mutations is unclear.
Here we investigated the phenotypes of three children with OI from two non-consanguineous families of Chinese Han origin, detected pathogenic gene mutations in TMEM38B and prospectively observe the effects of BP, zoledronic acid on bone of these patients.
Materials and methods
Subjects
Three Chinese patients from two families were diagnosed with OI in the Department of Endocrinology of the Peking Union Medical College Hospital between 2012 and 2014. The pedigrees of these two families were shown in Figure 1. The study protocol was approved by the Ethic Committee of Peking Union Medical College Hospital, and signed informed consents were obtained from the parents of the patients before their participation in this study.

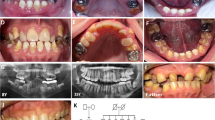
Pedigree of the osteogenesis imperfecta families in this study. The proband was designated with an arrow. Sanger sequencing results of three patients and their parents were shown. In patient 1, a novel homozygous mutation in TMEM38B was identified as c.455-7T>G in intron 3. In patients 2 and 3, a novel homozygous mutation in TMEM38B was identified as c.507G>A in exon 4. All parents of the patients were asymptomatic heterozygous carriers for the mutations. In family 1, younger sister (II3) was asymptomatic heterozygous carrier of the mutation and elder sister of patient 1 (II1) did not carry the mutation.
Clinical evaluation
Clinical data, which included bone fracture history, growth and development, were investigated in detail. Fractures were determined by medical history and bone X-ray films. Height and weight of patients were measured with Harpenden stadiometer (Seritex Inc., East Rutherford, NJ, USA) at baseline and every 6 months. Age-adjusted and sex-adjusted height was calculated based on a growth curve of Chinese children. Blood biochemistry, including levels of total calcium (Ca), phosphate (P), total alkaline phosphatase (a bone formation marker), alanine aminotransferase and creatinine, was measured using an automatic biochemical analyzer at the central laboratory of Peking Union Medical College Hospital. Serum levels of β-cross-linked C-telopeptide of type I collagen (β-CTX; a bone resorption marker), 25-hydroxyvitamin D and intact parathyroid hormone were measured by an automated electrochemiluminescence system (Roche Diagnostics, Basel, Switzerland). BMD at lumbar spine (L2–L4), femoral neck and total hip was measured by dual-energy X-ray absorptiometry (Lunar Prodigy, Fairfield, CT, USA) with appropriate pediatric software.
Detection of pathogenic mutation by targeted next-generation sequencing
Peripheral blood samples were obtained from the three patients, their parents and sisters. Blood samples were also drawn from 100 unaffected, unrelated, healthy subjects to confirm that the gene mutations were not merely polymorphisms. Genomic DNA was extracted from white blood cells using the DNA Extraction Kit (QIAamp DNA; Qiagen, Frankfurt, Germany) according to the manufacturer’s instructions.
In order to identify the pathogenic genetic defect, genomic DNA of the three patients were sequenced using a targeted next-generation sequencing panel (Illumina HiSeq2000 platform, Illumina, Inc., San Diego, CA, USA) that covered 700 genes implicated in genetic bone disorders. Known pathogenic genes of OI were included in this panel, including COL1A1, COL1A2, IFITM5, PPIB, SERPINF1, CRTAP, LEPRE1, SERPINH1, FKBP10, SP7, BMP1, TMEM38B and WNT1. The overall sequencing coverage of the target regions was ⩾98.95% for a 200 × depth of coverage in each of the chromosomes. Analysis of variants was restricted to the coding and flanking intronic sequences. Variants were detected using SAMtools (version 1.4, Tianjin, China) and SOAPsnp software (version 2.0, Tianjin, China). We filtered out the variants that had a minor allele frequency (MAF) ⩾0.005 in the Single Nucleotide Polymorphism Database (dbSNP build 137), the 1000 Genomes Project, the National Heart, Lung, and Blood Institute (NHLBI), the Exome Sequencing Project Exome Variant Server, the UCSC common SNP database and an internal control database.18
We further analyzed only those variants expected to produce damaged protein, including frame shift variants, nonsense variants, missense variants, and acceptor and donor splice sites.19 Prediction of missense variants was performed using Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) software. Potential splice site variants were predicted with NNSPLICE0.9 (http://www.fruitfly.org/) and Human Splicing Finder (http://www.umd.be/HSF3/HSF.html).
Mutation confirmation by Sanger sequencing
To confirm the mutation, fragments covering the mutation site in TMEM38B identified by next-generation sequencing were amplified by PCR. Primers for PCR amplification were designed using Oligo 7.0 software (Colorado Springs, CO, USA), which were 5′-CACCCACATTTAAAGTCCCT-3′ for the forward primer and 5′-GTGATAGAATTATGGCTCCCT-3′ for the reverse primer. PCR was conducted with TaqMix DNA polymerase (Biomed, Beijing, China) and its standard buffer under the following conditions: initial denaturation at 95 °C for 3 min, followed by 35 cycles at 95 °C for 30 s, 51 °C for 30 s and 72 °C for 60 s. PCR products were purified and sequenced using an ABI 377 DNA automated sequencer with dye terminator cycle sequencing kits (Applied Biosystems, Foster, CA, USA). The abnormal DNA sequences were identified by comparison with the NCBI reference sequence (NM_018112.2).
Evaluation of the mRNA changes induced by the splicing mutation
To evaluate the changes of mRNA induced by the splicing mutation, reverse transcriptase–PCR amplification was performed. Total RNA was isolated from whole blood using the RNA Extraction Kit (QIAamp RNA Blood Mini Kit; Qiagen) according to the manufacturer’s instructions. Total RNA was reverse-transcribed to generate complementary DNA (cDNA) using the Goscript Reverse Transcription system kit (Promega, Madison, WI, USA). Specific primer for PCR amplification of TMEM38B cDNA was designed, which were 5′-ATTCTCCATGGGACGAGTTG-3′ for the forward primer and 5′-GCCATTGGAAGGTGACTTTG-3′ for the reverse primer. PCR was performed using the methods similar to above mentioned.
Results
Clinical features
The phenotypes of the patients were presented in Table 1. The patients had no family history of bone fractures.
Family 1
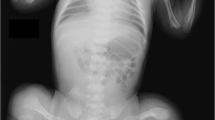
Patient 1 was a 4.5-year-old girl from a healthy non-consanguineous family. She was born full-term with a birth weight of 3250 g. She suffered from a femoral fracture 8 days after birth. Since then, she sustained five bilateral femur fractures after minor injuries. Intellect and milestones of development were normal. The family history was non-contributory. She went to our clinic at the age of 3.5 years. Her height was 93 cm (3rd to 10th percentile) and her weight was 15 kg (25th to 50th percentile). Physical examination revealed bilateral bowing femurs and blue sclera, without signs of abnormal hearing, teeth, joints or skin (Figure 2). Serum levels of calcium, phosphate, alkaline phosphatase and parathyroid hormone were within age-matched normal ranges, whereas the level of the bone resorption marker (β-CTX) was higher than normal (1.2 ng ml−1). Radiological assessment demonstrated generalized osteoporosis, slender long bone with thin cortices, metaphyseal enlargement of distal femur and slight bone deformity. BMD Z-score was lower compared with healthy control of the same age and sex. Treatment with intravenous zoledronic acid infusions was started at 4 years of age; areal BMD Z-score increased but fractures still happened.

Phenotypes of the three osteogenesis imperfecta patients with TMEM38B mutations. (a–h) For patient 1: (a) no wormian bones at the occipital region; (b, c) slight scoliosis and osteoporosis without vertebral compression fractures; (d) bowing of femur with fractures and metaphyseal enlargement of distal femur; and (e–h) normal teeth, blue sclera and severe bowing of right femur. (i–o) For patients 2 and 3: (i) patient 2 and (m) patient 3. No wormian bones at the occipital region: (j, k) patient 2 and (n) patient 3. Osteoporosis without vertebral compression fractures and no scoliosis: (l) patient 2 and (o) patient 3. Zebra line at metaphysis and slender long bone with thin cortices: (p) short stature without facial features and severe bone deformity.
Her two sisters were 7.5- and 3-year-old, respectively, with no history of bone fracture.
Family 2
Patients 2 and 3, who were brothers, were 8- and 7-year-old boys from a non-consanguineous family, born full-term, with birth weights of 3400 and 2800 g. They first sustained femoral fractures when they started walking, and subsequently experienced six and two femoral fractures, respectively. Their parents had no history of fractures. The patients were diagnosed with OI when they were 5 and 4 years old. For patient 2, height and weight were 110 cm (25th to 50th percentile) and 20 kg (75th to 90th percentile), respectively. For patient 3, height and weight were 95 cm (<3rd percentile) and 21 kg (>97th percentile), respectively. They had no signs of abnormal sclera, hearing, teeth, skin or joints (Figure 2). Patients 2 and 3 had normal serum levels of calcium, phosphate and parathyroid hormone. However, patient 2 had increased serum levels of alkaline phosphatase (394U l−1) and β-CTX (1.6 ng ml−1). Patient 3 had normal levels of alkaline phosphatase (263U l−1) and elevated levels of β-CTX (1.3 ng ml−1). Radiological assessment demonstrated generalized osteoporosis and slender long bone with thin cortices. Wormian bones and multiple vertebral compression fractures were absent (Figure 2). BMD Z-score was lower compared with healthy control of the same age and sex. Treatment with intravenous infusions of zoledronic acid was started at 6 years of age in patient 2 and at 5 years of age in patient 3, BMD Z-score increased and fractures reduced during 2-year follow-up.
Mutations of TMEM38B
A novel homozygous single-base-pair variation in TMEM38B (c.455-7T>G), located seven bases upstream of exon 4, was identified in patient 1 (Figure 1). The variation was predicted to create a new acceptor splice site with splice scores of 0.99 (NNSPLICE0.9) and 81.7 (Human Splicing Finder). The consequence of the mutation on splicing was further confirmed at mRNA level. The new splicing site induced the insertion of six bases (AGTTTT) between exons 3 and 4, causing an insertion of two amino acids (p.R151_G152insVL) (Figure 3b). The parents and younger sister (II3) of patient 1 were asymptomatic heterozygous carriers of the variation (Figure 1). Her elder sister (II1) was completely normal who did not carry the mutation. The affected TMEM38B sequence was highly conserved across species (Figure 3c).

The effect of point mutation (c.455-7T>G) in intron 3 on TMEM38B splicing. (a) Schematic view of TMEM38B exon and intron structure. (b) Sanger sequencing results of the reverse transcriptase–PCR products of patient 1 and a normal control were shown. 455-7T>G in intron 3 induced the insertion of six bases (AGTTTT) between exons 3 and 4. (c) R151 and G152 were highly conserved among 10 different species.
A novel homozygous mutation, a c.507G>A transition in exon 4 of TMEM38B, was identified in patients 2 and 3. The mutation led to the generation of a new downstream termination codon (p.W169X). The parents of these patients were heterozygous carriers of the mutation (Figure 1).
The TMEM38B mutations identified in the patients were absent from the 100 healthy controls, and did not match polymorphisms in any of the public databases. No mutations were identified in other genes that were known to be associated with dominant or recessive OI.
Discussion
Mutations of TMEM38B were recently reported to lead to a rare autosomal recessive form of OI. We report, for the first time, mutations in TMEM38B in patients with OI in Chinese population. Three patients presented with early-onset recurrent fractures, low bone mass, mild bone deformity and growth retardation, but no dentinogenesis imperfecta and hearing loss. We identified two novel mutations (c.455-7T>G in intron 3 and c.507G>A in exon 4) in TMEM38B in three Chinese OI patients through targeted next-generation sequencing, and confirmed their parents were heterozygous carriers.
The mechanism of mutation in TMEM38B leading to OI has not been completely elucidated. TRIC was first discovered in 2007,11 of which two isoforms had been identified in human and mice: TRIC-A, which was predominantly expressed in the sarcoplasmic reticulum of excitable tissues such as striated muscle and brain; and TRIC-B, which was expressed in the ER of most mammalian tissues.20, 21, 22 TRIC-B behaves as a K+ channel, acting to maintain intracellular calcium release in non-excitable cells. TRIC-B−/− neonatal mice were cyanotic and died shortly after birth, due to respiratory failure. However, the skeletal phenotype was not described in TRIC-B−/−mice.14, 15, 20, 21 Recently, OI with TMEM38B mutations were ascribed to collagen modification defects because multiple proteins in the ER involved in collagen metabolism were calcium-dependent. TMEM38B mutations could alter post-translational modification of collagen because of its role in modulating interaction of calreticulin with cyclophilin B and with protein disulfide isomerase.23 In addition, reduced calcium release induced by TMEM38B mutations would inactivate Ca2+-dependent enzymes through Ca2+-dependent kinase/phosphatase signaling, therefore, the conversion of multipotent mesenchymal cells into osteoblasts was inhibited.24, 25, 26 Abnormal Ca2+ handling could cause rapid reorganization of the ER membrane and activate the unfolded protein response, leading to ER stress.27, 28, 29 It was consistent with the swollen ER structures observed in airway epithelial cells in TRIC-B−/− mice.15 Severe and prolonged ER stress could lead to cytotoxicity, induce osteoblast apoptosis and affect secretion of bone matrix. Therefore, several alterations in intracellular or extracellular metabolic pathways may be involved in pathogenesis of OI induced by TMEM38B mutations.
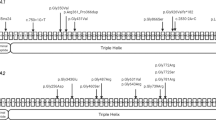
Four mutations in TMEM38B had been reported in OI patients, including our findings in this study. Previous study indicated that the c.455_542del mutation led to an abnormal transcription without exon 4, and created a premature stop codon (p.Gly152Alafs*5).11, 12 An approximately fourfold reduction in TMEM38B mRNA levels was found in lymphoblastoid cells in individuals with c.455_542del mutation, which suggested an occurrence of nonsense-mediated decay.12 SMART web-based analysis implies that the premature stop codon leads to a deletion of nearly one-third of the TRIC domain, the protein’s functional domain.11, 12 The 35-kb deletion involving exons 1 and 2 of TMEM38B could produce protein with aberrant TM1 and pore regions, which were important for ion conduction.12, 30 We identified two novel homozygous mutations in the TMEM38B gene (c.455-7T>G and c.507G>A) in our OI patients, both of which were point mutations and were different from previous identified mutations. The c.455-7T>G mutation was a splicing site mutation, which led to an insertion of two amino acids (p.R151_G152insVL). As R151 and G152 were highly conserved across species, we assumed the insertion of VL would influence the function of TIRC-B. In the topology model, R151_G152insVL affected TM2, a major pore-lining domain, which was extensively involved in channel gating.31 The c.507G>A mutation in exon 4 could induce a premature stop codon and a truncated protein. We assumed that the severe functional abnormalities in aberrant protein caused by c.507G>A mutation in exon 4 might potentially affected TM3 and the lumen region near TM2 in the topology model (Figure 4). As the exact function of TMEM38B and its domains are not completely clear, the mechanisms by which mutations in TMEM38B lead to OI still require to be investigated further.

Topology model of trimeric intracellular cation channels on the sarcoplasmic reticulum/endoplasmic reticulum (SR/ER) membrane. The c.455-7T>G mutation in intron 3 affected TM2. The c.507G>A mutation in exon 4 affected TM3 and the lumen region near TM2. The c.455_542del mutation in exon 4 affected TM3, TM2, and the lumen region nearby. The 35kb deletion involving exons 1 and 2 affected TM1 and the pore region.
The phenotypes of OI with TMEM38B mutations were reported in 22 patients.11, 12, 13 Patients with TMEM38B mutations had various fracture frequency, mild to moderate short stature. Mildly gray-blue sclera was described in several children. Only one child had hearing impairment and no patients had tooth defects.13 The phenotypes of our patients were similar to those reported previously (Table 2). Our patients suffered from 3–7 fractures under minor trauma. One of our patients had blue sclera, but the others had no extra-skeletal abnormalities. It was difficult to establish a genotype–phenotype correlation because of the limitation of the sample size. In our patients, zoledronic acid increased BMD, reduced bone turnover biomarker levels and with good tolerance. In an Albanian girl with a 35-kb deletion in TMEM38B, fracture incidence was reduced after treatment with neridronate.13 The efficacy of bisphophonate treatment in OI patients with TMEM38B mutations still need to be confirmed in a large sample of patients.
In conclusion, TMEM38B mutations could lead to a rare form of OI, with an autosomal recessive pattern of inheritance. We identified two novel mutations (c.455-7T>G in intron 3 and c.507G>A in exon 4) in TMEM38B in three Chinese children with OI. The two mutations created a new acceptor splice site (p.R151_G152insVL) and a novel downstream termination codon (p.W169X), respectively. These patients presented moderate OI phenotypes, with early-onset multiple fractures, mild skeletal deformities and growth retardation. Our findings of the novel mutations in TMEM38B expand the pathogenic spectrum of OI and strengthen the role of TRIC-B in the pathogenesis of OI.
References
Forlino, A., Cabral, W. A., Barnes, A. M. & Marini, J. C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 7, 540–557 (2011).
Cundy, T. Recent advances in osteogenesis imperfecta. Calcif. Tissue Int. 90, 439–449 (2012).
Van Dijk, F. S. & Sillence, D. O. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A 164A, 1470–1481 (2014).
Kim, O. H., Jin, D. K., Kosaki, K., Kim, J. W., Cho, S. Y., Yoo, W. J. et al. Osteogenesis imperfecta type V: clinical and radiographic manifestations in mutation confirmed patients. Am. J. Med. Genet. A 161, 1972–1979 (2013).
Laine, C. M., Wessman, M., Toiviainen-Salo, S., Kaunisto, M. A., Mayranpaa, M. K., Laine, T. et al. A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J. Bone Miner. Res. 30, 510–518 (2015).
Garbes, L., Kim, K., Rieß, A., Hoyer-Kuhn, H., Beleggia, F., Bevot, A. et al. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am. J. Hum. Genet. 96, 432–439 (2015).
Mendoza-Londono, R., Fahiminiya, S., Majewski, J., Care4Rare Canada Consortium, Tetreault, M., Nadaf, J. et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am. J. Hum. Genet. 96, 979–985 (2015).
Symoens, S., Malfait, F., D’hondt, S., Callewaert, B., Dheedene, A., Steyaert, W. et al. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J. Rare Dis. 8, 154 (2013).
Rauch, F., Fahiminiya, S., Majewski, J., Carrot-Zhang, J., Boudko, S., Glorieux, F. et al. Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am. J. Hum. Genet. 96, 425–431 (2015).
Bonafe, L., Cormier-Daire, V., Hall, C., Lachman, R., Mortier, G., Mundlos, S. et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. A. 167, 2869–2892 (2015).
Shaheen, R., Alazami, A. M., Alshammari, M. J., Faqeih, E., Alhashmi, N., Mousa, N. et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J. Med. Genet. 49, 630–635 (2012).
Volodarsky, M., Markus, B., Cohen, I., Staretz-Chacham, O., Flusser, H., Landau, D. et al. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum. Mutat. 34, 582–586 (2013).
Rubinato, E., Morgan, A., D’Eustacchio, A., Pecile, V., Gortani, G., Gasparini, P. et al. A novel deletion mutation involving TMEM38B in a patient with autosomal recessive osteogenesis imperfecta. Gene 545, 290–292 (2014).
Yazawa, M., Ferrante, C., Feng, J., Mio, K., Ogura, T., Zhang, M. et al. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 448, 78–82 (2007).
Yamazaki, D., Komazaki, S., Nakanishi, H., Mishima, A., Nishi, M., Yazawa, M. et al. Essential role of the TRIC-B channel in Ca2+ handling of alveolar epithelial cells and in perinatal lung maturation. Development 136, 2355–2361 (2009).
Colquhoun, D. & Sivilotti, L. G. Function and structure in glycine receptors and some of their relatives. Trends Neurosci. 27, 337–344 (2004).
Dwan, K., Phillipi, C. A., Steiner, R. D. & Basel, D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst. Rev. 7 (2014).
Sule, G., Campeau, P. M., Zhang, V. W., Nagamani, S. C., Dawson, B. C., Grover, M. et al. Next-generation sequencing for disorders of low and high bone mineral density. Osteoporos. Int. 24, 2253–2259 (2013).
El Shamieh, S., Boulanger-Scemama, E., Lancelot, M. E., Antonio, A., Demontant, V., Condroyer, C. et al. Targeted next generation sequencing identifies novel mutations in RP1 as a relatively common cause of autosomal recessive rod-cone dystrophy. BioMed Res. Int. 2015, 485624 (2015).
Venturi, E., Sitsapesan, R., Yamazaki, D. & Takeshima, H. TRIC channels supporting efficient Ca(2+) release from intracellular stores. Pflugers Arch. 465, 187–195 (2013).
Zhou, X., Lin, P., Yamazaki, D., Park, K. H., Komazaki, S., Chen, S. R. et al. Trimeric intracellular cation channels and sarcoplasmic/endoplasmic reticulum calcium homeostasis. Circ. Res. 114, 706–716 (2014).
Yamazaki, D., Tabara, Y., Kita, S., Hanada, H., Komazaki, S., Naitou, D. et al. TRIC-A channels in vascular smooth muscle contribute to blood pressure maintenance. Cell Metab. 14, 231–241 (2011).
Forlino, A. & Marini, J. C. Osteogenesis imperfecta. Lancet (e-pub ahead of print 2 November 2015; doi:10.1016/S0140-6736(15)00728-X.
Chen, G., Deng, C. & Li, Y. P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 8, 272–288 (2012).
Bodine, P. V. & Komm, B. S. Wnt signaling and osteoblastogenesis. Rev. Endocr. Metab. Disord. 7, 33–39 (2006).
Zayzafoon, M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J. Cell. Biochem. 97, 56–70 (2006).
Varadarajan, S., Tanaka, K., Smalley, J. L., Bampton, E. T., Pellecchia, M., Dinsdale, D. et al. Endoplasmic reticulum membrane reorganization is regulated by ionic homeostasis. PLoS ONE 8, e56603 (2013).
Wu, Y., Yang, M., Fan, J., Peng, Y., Deng, L., Ding, Y. et al. Deficiency of osteoblastic Arl6ip5 impaired osteoblast differentiation and enhanced osteoclastogenesis via disturbance of ER calcium homeostasis and induction of ER stress-mediated apoptosis. Cell Death Dis. 5, e1464 (2014).
Luciani, D. S., Gwiazda, K. S., Yang, T. L., Kalynyak, T. B., Bychkivska, Y., Frey, M. H. et al. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 58, 422–432 (2009).
Wollmuth, L. P. & Sobolevsky, A. I. Structure and gating of the glutamate receptor ion channel. Trends Neurosci. 27, 321–328 (2004).
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K. et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 (2010).
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81100623 and No. 8157041048) and National Key Program of Clinical Science (WBYZ2011-873). We thank the staff in the Department of Radiology for measurement of bone mineral density and interpretation of X-ray films for the patients with OI. We also thank all patients with TMEM38B mutations and their families for their participation in this research and thank all unaffected, unrelated individuals for providing control DNA samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Lv, F., Xu, Xj., Wang, Jy. et al. Two novel mutations in TMEM38B result in rare autosomal recessive osteogenesis imperfecta. J Hum Genet 61, 539–545 (2016). https://doi.org/10.1038/jhg.2016.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.11
This article is cited by
-
Mesenchymal stem cells in the treatment of osteogenesis imperfecta
Cell Regeneration (2023)
-
Atypical cell death and insufficient matrix organization in long-bone growth plates from Tric-b-knockout mice
Cell Death & Disease (2023)
-
Novel splice site variant of TMEM38B in osteogenesis imperfecta type XIV
Human Genome Variation (2023)
-
Novel Mutations in PLOD2 Cause Rare Bruck Syndrome
Calcified Tissue International (2018)
-
Ion- and water-binding sites inside an occluded hourglass pore of a trimeric intracellular cation (TRIC) channel
BMC Biology (2017)
