
Abstract
Ichthyosis follicularis with atrichia and photophobia (IFAP) syndrome is a rare genetic disorder characterized by the triad of ichthyosis follicularis, alopecia and photophobia. Previous studies have identified five missense mutations in the membrane-bound transcription factor protease, site 2 (MBTPS2) gene in European patients with this syndrome. In this study, we detected the 1286G>A (Arg429His) mutation in MBTPS2 in a Japanese patient with IFAP syndrome. This mutation has previously been detected in a German family with this syndrome. Functional analysis revealed that this mutation was the most severe mutation identified to date for this syndrome. None of the male German patients had been tested for the mutation because they had several visceral and bone anomalies, and had died as neonates or infants. The clinical features of our 5-year-old patient are less severe than those of the German patients. Although he has neurological abnormalities, such as retarded psychomotor development and seizures, he does not have bone or visceral anomalies, except cryptorchidism. This case indicates the existence of other factor(s) that influence the clinical features of this syndrome. Further clinical and genetic studies are required to clarify the relationship between phenotypes and genotypes and to identify such modifying factors.
Similar content being viewed by others

Introduction
Ichthyosis follicularis with atrichia and photophobia (IFAP) syndrome (MIM 308205) is a rare congenital disorder characterized by generalized ichthyotic skin changes with follicular hyperkeratosis, congenital hairlessness and photophobia, as well as additional clinical findings.1 X-linked recessive transmission has been suggested for this syndrome because most patients are male,2 and the full phenotype is found only in males.3, 4 Oeffner et al.5 performed a linkage analysis using two families of European descent, in which IFAP segregated according to an X-linked pattern of transmission. They identified five missense mutations in the membrane-bound transcription factor protease, site 2 gene (MBTPS2; MIM 300294) encoding a membrane-embedded zinc metalloprotease that activates signaling proteins involved in the endoplasmic reticulum stress response and in the sterol control of transcription.5 In this study, we report the case of a Japanese patient with the IFAP triad, short stature, mental retardation and seizures. The MBTPS2 Arg429His mutation, which was previously identified by Oeffner et al.5 in male patients most severely affected by this syndrome, was detected in this patient.
Case report
The patient was a 2-year-old male child who was born to healthy non-consanguineous parents and was referred to our institution for seizures and severe mental and growth retardation. Ultrasonography performed at 21 weeks of gestation revealed fetal intrauterine growth retardation, and delivery occurred at 37 weeks. The birth weight of the patient was 2167 gm and height was 51.5 cm; he lacked scalp hair, eyebrows and eyelashes, and exhibited generalized ichtyosis. At 3 months of age, his serum total immunoglobulin E level was 4945 IU ml−1, and his serum specific immunoglobulin E levels to albumen and milk were elevated. The patient also had bilateral cryptorchidism, which required surgery. Photophobia became apparent during the first year of life. By 1 year of age, he experienced a brief generalized tonic–clonic seizure with high fever. Later myoclonic seizures appeared without fever.
On admission, his weight, height and head circumference were all below the third percentile, and bone age was below chronologic age. Physical examination revealed that there was no visceromegaly, and cardiovascular examination yielded normal results. The patient did not have scalp hair, eyebrows and eyelashes (Figure 1). He exhibited generalized skin dryness, which led to severe itching; eczematous changes in the arms and legs; and thickened nails. He had normal teeth. Detailed pathological analysis of the skin biopsy has been reported previously.6 Ophthalmological evaluation revealed severe photophobia and bilateral corneal vascularization. Neurological examination revealed that the patient was alert but mentally retarded. The patient could smile and visually follow faces but could not talk. He was able to sit unassisted but could not walk. His cranial nerves were unaffected; although a slight decrease in muscle tone was observed, the muscle stretch reflexes were normal. Normal plantar responses and withdrawal to painful stimulation were observed. The results for routine hematological screening tests; liver and thyroid function profiles; plasma amino acid levels; urinalysis; visual- and brainstem auditory-evoked potentials; electrocardiography; and chest, skull and spine radiography were normal or negative. Magnetic resonance imaging revealed enlargement of the cisterna magna and irregular deficiency in the medial occipital lobe seems as schizencephaly, and irregular distortion on the anterior horn of the lateral ventricle, which expanded in the lateral superior direction. Electroencephalography revealed multifocal localization spike on the right central, right parietal and left occipital regions. Spikes were observed on the right parietal region even after antiepileptic therapy. The parents and the younger brother of the patient did not exhibit any of these clinical features.

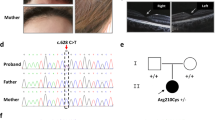
Photograph of the patient, showing alopecia and absence of eyebrows and eyelashes.
Currently (age, 5 years 4 months), his weight, height and head circumference are 10.4 kg (<third centile), 88.1 cm (<third centile) and 44.5 cm (<third centile), respectively. He can walk using a walk aid and speak a few words. The seizures were effectively controlled using valproic acid, diazepam and zonisamide. He develops urticaria on exposure to peanuts and tree nuts (that is, hazelnuts and walnuts).
Molecular and cytogenetic studies
Blood samples were collected from the patient and his family after having obtained written and informed consent from unaffected family members according to a protocol reviewed and approved by the ethical committee of the University of Tsukuba. Chromosomal analysis revealed that the patient had a normal karyotype, 46, XY. Sequence analysis of MBTPS2 was performed according to a previously reported method.5 We identified a missense mutation, c.1286G>A, that caused a Arg429His substitution in the patient. This mutation was found in the proband and his unaffected mother but not in his unaffected brother (Figure 2).

Family pedigree and mutation analysis. (a) Pedigree of the family studied. The circle indicates female individuals, and squares indicate male individuals. The filled symbol denotes the affected individual, and the dot with symbol denotes a carrier individual. An arrow indicates the proband. (b) Sequence analysis of MBTPS2. 1286G>A (Arg429His) mutation was identified in the proband (P) and his mother (M) but not in his brother (B).
Discussion
We detected a Arg429His mutation in MBTPS2 in a Japanese patient with IFAP syndrome. This is the first case, in which an MBTPS2 mutation has been identified in a Japanese patient with this syndrome.
The IFAP syndrome was first identified by MacLeod et al.1 in three brothers in 1909. It is a rare X-linked genetic disorder, and <30 cases have been reported. Such patients have a unique appearance because of the alopecia, photophobia and generalized follicular hyperkeratosis. Inconsistent findings include neurological abnormalities, such as retarded psychomotor development, cerebral atrophy, temporal lobe malformation, hypoplasia of the corpus callosum and seizures; failure to thrive; nail dystrophy; atopic manifestations; inguinal hernia; aganglionic megacolon; as well as renal, vertebral and testicular anomalies.7, 8, 9, 10, 11, 12
Oeffner et al.5 reported that IFAP syndrome is caused by functional deficiency of membrane-bound transcription factor protease, site 2, an intramembrane zinc metalloprotease that is essential for cholesterol homeostasis and the ER stress response.13, 14, 15 They performed a linkage analysis involving two families of European descent, in which IFAP segregated according to an X-linked pattern of transmission, and assigned the IFAP locus to the 5.4-Mb region between DXS989 and DXS8019 on Xp22.11–p22.13. They identified five missense mutations exchanging highly conserved amino acids in MBTPS2 at Xp22.11 in five unrelated patients of European descent.
Our patient has a missense mutation, 1286G>A, leading to an Arg429His substitution in MBTPS2. The same mutation has previously been reported in a German family with IFAP syndrome.5 The manifestations of two male patients from this family have been compared with those of our patient in Table 1. In this family, three female patients with skin manifestations (that is, dry skin, and atrophoderma with linear lesions) and two unaffected female patients carried this mutation heterozygously. This family included four male patients; however, they had not been tested because they had several visceral and bone anomalies and had died within 2 years after birth. One male patient was a collodion baby who also had motor retardation, a cleft palate, unilateral cleft hand, two butterfly vertebrae, bilateral inguinal hernia, omphalocele, stenosis of the small intestine and Hirschsprung disease; he lacked one kidney. Another male patient had microcephaly, an arachnoid cyst, Arnold–Chiari malformation type I, thoracolumbar hydromyelia, seizures, psychomotor retardation, retrognathia, deficient growth, cleft hands, a butterfly vertebra, a wedge-shaped vertebra, an atrial septal defect, arterial hypertension, recurrent infection of the upper airways, hypospadias, choanal stenosis, inguinal hernia and Hirschsprung disease; he lacked one kidney.
Oeffner et al.5 suggested that missense mutations in MBTPS2 are responsible for the IFAP phenotype and that the degree of clinical severity correlates with the reduction in activity.They tested the effect of the five MBTPS2 missense mutations detected in IFAP syndrome patients on the potential to complement S2P deficiency in Chinese hamster CHO-K1-M19 cells and on the stimulation of sterol-responsive elements luciferase reporter. M19 cells in which the orthologous Mbtps2 is deleted cannot grow in cholesterol and lipid-deficient culture media.13 Wild-type MBTPS2 stably transfected into the M19 cells complemented this defect and restored their wild-type growth characteristics. None of the five mutants detected in IFAP patients retained this property to the same extent as did the wild-type gene; however, great variation was observed in residual activity. In mutant Arg429His, almost no survival was detected.5 A luciferase reporter gene under transcriptional control of sterol-responsive elements was active in cells grown in sterol-deficient media on cotransfection with wild-type MBTPS2. However, sterol responsiveness of the cells transfected with the mutants was restored to a lesser extent than that in cells transfected with wild-type MBTPS2 and also differed considerably among the mutants. The Arg429His mutation had the lowest residual activity.5 Thus, the Arg429His mutation is considered as the most severe MBTPS2 mutation till date. Our patient is now 5 years old, and his clinical features are much less severe than those of the German patients reported by Oeffner et al. Although he has neurological abnormalities, such as retarded psychomotor development and seizures, he does not have bone or visceral anomalies, except cryptorchidism.
Further clinical and genetic studies are required to clarify the relationship between phenotypes and genotypes and to identify any additional factor(s) that may have a role in the pathogenesis of IFAP spectrum disorders.
References
Macleod, J. M. H. Three cases of ‘ichthyosis follicularis’ associated with baldness. Br. J. Dermatol. 21, 165–189 (1909).
Hamm, H., Meinecke, P. & Traupe, H. Further delineation of the ichthyosis follicularis, atrichia, and photophobia syndrome. Eur. J. Pediatr. 150, 627–629 (1991).
Eramo, L. R., Esterly, N. B., Zieserl, E. J., Stock, E. L. & Herrmann, J. Ichthyosis follicularis with alopecia and photophobia. Arch. Dermatol. 121, 1167–1174 (1985).
König, A. & Happle, R. Linear lesion reflecting lionization in women heterozygous for IFAP syndrome (ichthyosis follicularis with atrichia and photophobia). Am. J. Med. Genet. 85, 365–368 (1999).
Oeffner, F., Fischer, G., Happle, R., König, A., Betz, R.C., Bornholdt, D. et al. IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am. J. Hum. Genet. 84, 459–467 (2009).
Kamo, M., Ohyama, M., Shimizu, T., Kosaki, K., Amagai, M., Ebihara, T. et al. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome: a case report and a pathological insight into pilosebaceous anomaly. Am. J. Dermatopathol. (in press).
Martino, F., D’Eufemia, P., Pergola, M. S., Finocchiaro, R., Celli, M., Giampà, G. et al. Child with manifestations of dermotrichic syndrome and ichthyosis follicularis-alopecia-photophobia (IFAP) syndrome. Am. J. Med. Genet. 44, 233–236 (1992).
Keyvani, K., Paulus, W., Traupe, H., Kiesewetter, F., Cursiefen, C., Huk, W. et al. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome: Clinical and neuropathological observationsin a 33-year-old man. Am. J. Med. Genet. 78, 371–377 (1998).
Boente, M. C., Bibas-Bonet, H., Coronel, A. M. & Asial, R. A. Atrichia, ichthyosis, follicular hyperkeratosis, chronic candidiasis, keratitis seizures, mental retardation and inguinal hernia: a severe manifestation of IFAP syndrome? Eur. J. Dermatol. 10, 98–102 (2000).
Bibas-Bonet, H., Fause, R., Boente, M. C., Coronel, A. M. & Asial, R. IFAP syndrome ‘plus’ seizures, mental retardation, and callosal hypoplasia. Pediatr. Neurol. 24, 228–231 (2001).
Mégarbané, H., Zablit, C., Waked, N., Lefranc, G., Tomb, R. & Mégarbané, A. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome: report of a new family with additional features and review. Am. J. Med. Genet. A 124, 323–327 (2004).
Happle, R. What is IFAP syndrome? Am. J. Med. Genet. A 124, 328 (2004).
Rawson, R. B., Zelenski, N. G., Nijhawan, D., Ye, J., Sakai, J., Hasan, M. T. et al. Complementation cloning of SP2, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell. 1, 47–57 (1997).
Zelenski, N. G., Rawson, R. B., Brown, M. S. & Goldstein, J. L. Membrane topology of SP2, a protein required for intramembranous cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 274, 21973–21980 (1999).
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Dave, U. P., Prywes, R. et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 6, 1355–1364 (2000).
Acknowledgements
We are grateful to the proband and his family for their participation in the study. This work was supported by Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Sciences (21591331).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakayama, J., Iwasaki, N., Shin, K. et al. A Japanese case of ichthyosis follicularis with atrichia and photophobia syndrome with an MBTPS2 mutation. J Hum Genet 56, 250–252 (2011). https://doi.org/10.1038/jhg.2010.163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.163
Keywords
This article is cited by
-
MBTPS2, a membrane bound protease, underlying several distinct skin and bone disorders
Journal of Translational Medicine (2021)
-
Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome
Orphanet Journal of Rare Diseases (2011)
