
Abstract
A54145 is a complex of lipopeptide antibiotics produced by Streptomyces fradiae. A54145 factors are structurally related to daptomycin, with four modified amino acids, only one of which is present in daptomycin. We generated three mutants defective in lptJ, lptK or lptL, whose gene products are involved in the formation of hydroxy-Asn3 (hAsn3) and methoxy-Asp9 (moAsp9). Each of the mutants produced novel lipopeptides related to A54145 and the profiles allowed assignment of functions for those genes. We constructed strains carrying different combinations of these genes coupled with a mutation in the lptI gene involved in the biosynthesis of 3-methyl-Glu12 (3mGlu12), and all recombinants produced novel lipopeptides. One of the compounds displayed very good antibacterial activity in the presence of bovine surfactant, which interacts with daptomycin or A54145E to inhibit their antibacterial activities.
Similar content being viewed by others
Introduction
A54145 (Figure 1) is a complex of calcium-dependent lipodepsipeptide antibiotics produced by Streptomyces fradiae NRRL 18160.1, 2 A54145 has a cyclic depsipeptide ring containing 10 amino acids and an exocyclic tail of three amino acids, and it is similar in overall structure to daptomycin. Like A21987C and daptomycin,3, 4 the N-terminal Trp1 of the exocyclic peptide is coupled to long-chain length fatty acids, the most common being iso-decanoyl, n-decanoyl and anteiso-undecanoyl in A54145 factors.1, 2 A54145 factors also vary at positions 12 and 13, in which different factors have different combinations of Glu12, L-3-methyl-Glu12 (3mGlu12), Ile13, or Val13. The most prevalent factors produced during the S. fradiae fermentation are shown in Figure 1. Typically, A54145 factors containing Glu12 accumulate early in fermentation and those containing 3mGlu12 accumulate later, with final yields of about 60% of factors containing Glu12.1 The study by Boeck et al.5 demonstrated that the distribution of A54145 factors can be manipulated by adding certain amino acids or lipids to the fermentation. For instance, feeding L-Ile, enriched for factors containing Ile13 from 89 to 98%, whereas feeding L-Val (which inhibited overall lipopeptide production by about 70%) enriched for compounds containing Val13 from 11 to 56%, almost exclusively in factor F (containing Glu12). Feeding either of these amino acids also dramatically shifted the distribution of lipid side chains attached to the core.

The structure of the A54145 factors normally produced by S. fradiae. Amino acid modifications to the lipopeptide that are of interest to this study are highlighted in gray.
The fatty-acid side chains of the A54145 factors can be removed by a deacylase produced by Actinoplanes utahensis,6 thus providing a means to generate the peptide core as a starting material for chemical acylations with different fatty acids.7 Several semisynthetic derivatives were tested for antibacterial properties and acute toxicity in mice.8 The most potent natural factors (B, B1 and E), contain 3mGlu12 and Ile13, but the LD50 for A54145B is low at only 28 mg kg−1. The natural factors containing Glu12 and Ile13 had about twofold lower antibacterial activities, but were substantially less toxic (for example, LD50>500 mg kg−1 for A54145A). A semisynthetic derivative of the A54145A core peptide containing an undecanoyl side chain had improved antibacterial activities in vitro (MICs ranging from 0.5 to 4 μg ml−1 for different Staphylococcal and Streptococcal strains), in vivo efficacy against Streptococcus pyogenes in a mouse protection assay (ED50=1.6 mg kg−1), and low toxicity (LD50>500 mg kg−1).8
Daptomycin has been approved for the treatment of complicated skin and skin structure infections caused by Gram-positive bacteria,9 and for the treatment of bacteremia and right-sided endocarditis caused by Staphylococcus aureus, including strains resistant to methicillin (MRSA).10 One shortcoming of daptomycin is its lack of efficacy in community-acquired pneumonia.11 The poor efficacy in lung tissue appears to be caused by the sequestration of daptomycin in the lung surfactant.12 To address this issue, derivatives of daptomycin were generated by modifying the lipid side chain or the δ-amino group of ornithine by medicinal chemistry,13, 14 and by modifying the core peptide by combinatorial biosynthesis.15, 16 Although improvements in antibacterial activity in the presence of surfactant relative to daptomycin were observed, none of the compounds were active enough to carry forward into clinical studies.
As part of the combinatorial biosynthesis program, the lpt gene cluster dedicated to the biosynthesis of A54145 was cloned and sequenced,17 the complete pathway was localized on a bacterial artificial chromosome (BAC) vector and expressed in heterologous streptomycete hosts.18 In addition, an ectopic transcomplementation system was developed to engineer the A54145 biosynthetic gene cluster in Escherichia coli using λ-Red-mediated recombination, conjugation from E. coli mediated by oriT from plasmid RP4, and site-specific integration of engineered plasmids into bacteriophage φC31 and φBT1 attB sites.18 Using this system, the A54145 nonribosomal peptide synthetase (NRPS) genes were genetically engineered to generate hybrid lipopeptides by module exchange.19 A mutant, defective in the lptI gene was engineered and confirmed that its gene product is the methyltransferase involved in the biosynthesis of 3mGlu12, resulting in the production of only the Glu12-containing factors.18 It is believed that LptI uses a similar mechanism, as the homologous GlmT methyltransferase from Streptomyces coelicolor converting α-ketoglutarate to the (2S,3R)-3-methylglutamate intermediate that is further converted to 3mGlu by a transaminase from primary metabolism.20 When fermented in DSF medium with isoleucine, the strain produced >0.5 g l−1 of A54145D.18
In addition to 3mGlu12, the A54145 factors have three other modified amino acids, L-hydroxy-Asn (hAsn3), sarcosine (Sar5) and L-methoxy-Asp (moAsp9). The biosynthesis of Sar is encoded in the lptA NRPS gene as a methyltransferase (M) domain that converts Gly to Sar, but the other amino acid modifications are believed to be catalyzed by individual oxygenases and an O-methyltransferase.17 In the present study, we constructed strains containing combinations of deletions of lptJ, lptK and lptL genes encoding the three enzymes predicted to be involved in the biosynthesis of hAsn and moAsp, and analyzed the lipopeptides produced during fermentation to confirm the gene functions. We combined these deletions with the lptI deletion, and with plasmids containing combinations of the lpt genes to generate a series of novel lipopeptides. The antibacterial activities of the A54145 derivatives were determined in the presence and absence of bovine pulmonary surfactant, of which, one compound displayed very good antibacterial activity.
Materials and Methods
Strains, plasmids, media, growth conditions and conjugation
The strains and plasmids used in this study are shown in Table 1. Luria-Bertani broth or Luria-Bertani agar21 was used for the growth of E. coli strains at 37°C, except when the temperature-sensitive plasmid pKD11922 was present. When appropriate, media were supplemented with ampicillin (Amp; 100 μg ml−1), apramycin (Am; 100 μg ml−1), chloramphenicol (Cm; 25 μg ml−1), tetracycline (Tc; 12.5 μg ml−1), neomycin (Nm; 50 μg ml−1), spectinomycin (Spc; 50 μg ml−1) or hygromycin B (Hm; 50 μg ml−1). The media and growth conditions for S. fradiae and the method for conjugation of plasmid DNA from E. coli to S. fradiae were as described.18 S. fradiae strains were grown in CSM broth or on mR2YE agar, and when appropriate supplemented with trimethoprim (Tmp; 50 μg ml−1), neomycin (10 μg ml−1), apramycin (10 μg ml−1) or hygromycin B (150 μg ml−1).
Truncation of the lpt cluster with to terminator-AmpR cassette
The to terminator-AmpR cassette was PCR amplified from pDA1836 and the λ-Red-mediated recombinations were carried out as described.18, 22 The PCR product was made using primers P1 (5′-GGGTAACGCCAGGGTTTTCCCAGTCACGACGTTGTAAAACGACGGCCATGTGTAGGCTGGAGCTGCTTC-3′, italicized letters indicate primer binding sites for pDA1836) and P2 (5′-CGCCCGCACCGACGAACTCCTGGCCGCCCACAAAGTGCTGTTCCTCAGAACGGATAACAATTTCACACAGGA-3′) to delete lptJ and downstream DNA by recombination from the integrative lpt cluster BAC pDA2002 to generate pDA2060. Additional PCR templates made in this manner were used to delete lptK (P1 and P3 (5′- TCGGGGCCAACATCGGCATGTTCACCCTCTTCGTACGCTCGGAACGATATATTAAGCGGATAACAATTTCACACAGGA-3′)) and lptL (P1 and P4 (5′-GGTCCCCCACCTCCCGGAGAAACAGCATGGAACCCGAGAACACCTTCACCCTGGATAACAATTTCACACAGGA-3′)), as well as downstream DNA, to generate pDA2074 and pDA2076, respectively, from pDA2002.
Construction of integrative, ermE*p containing BACs expressing multiple lpt genes
Derivatives of pCB01 with the φBT1 cassette and strong constitutive ermE* promoter inserted immediately upstream of the start codon of lptK or lptL were constructed by λ-Red-mediated recombination as described previously.18 The initial 0.3 kb of the lptK gene was PCR amplified from pCB01 using the primers P5 (5′-CTAGAATTCACATATGACCATCGCCCTCGC-3′, underlined letters indicate restriction sites) and P6 (5′-CAATCTAGACCGGATAGTGGGTGAACTCG-3′) and cloned adjacent to the φBT1 cassette-ermE* promoter to generate pDA1860. The ∼6 kb NheI fragment containing the BAC flanking region–φBT1 cassette-ermE*p::lptK was excised from the plasmid and used for λ-Red-mediated recombination into pCB01 to generate the BAC pJR2012. The resulting lptKLI expression plasmid was functionally validated by complementation of the DA1333 strain (ΔlptKLI) to generate DA1449.
Similarly, 0.3 kb from the start of the lptL gene was PCR amplified from pCB01 using the primers P7 (5′-AACCATATGGAACCCGAGAACACCTTCAC-3′) and P8 (5′-TGTTCTAGACGGGAAGGTTGCGCAGGAGC-3′) and cloned adjacent to the φBT1 cassette-ermE* promoter to generate pDA1862. The ∼6 kb fragment containing the flanking region–φBT1 cassette-ermE*p::lptL was excised from the plasmid with NheI digestion and used for λ-Red-mediated recombination into pCB01 to generate the BAC pJR2015. The lptLI expression plasmid was functionally validated by complementation of the DA1336 strain (ΔlptLI) to generate DA1621.
Construction of single lpt gene complementation plasmids
A DNA fragment containing the SpcR coding region was PCR amplified with the primers P9 (5′-CTAGAATTCACATATGAGGGAAGCGGTGATCG-3′) and P10 (5′-CAATCTAGAGTCTTCCCCAGCTCTCTAAC-3′) and ligated into NdeI and XbaI-digested ermE* promoter expression vector pDA1652,18 to generate pDA2112. The ermE*p::SpcR expression cassette flanked by the to and fd terminators was PCR amplified with primers P11 (5′-TCGGGTTAACTGCGGGGTCGTCAAC-3′) and P12 (5′-TGAGCGGCCGCAAGCTTACTGAGCAACGCGAAGGC-3′) and digested with HpaI and NotI. The digested PCR product was inserted into pRT802,23 digested with NotI and EcoRV to create pDA2113.
The to terminator-CmR cassette was generated by replacing the AmpR gene in pDA1836.18 with the CmR gene to generate pDA1842. The CmR gene was PCR amplified from pCB01 using the primers P13 (5′-ATCGCGGCCGCACGGAAGATCACTTC-3′) and P14 (5′-CGCAAGCTTCGCCGTCGACCAATTCTCATG-3′), digested with NotI and HindIII and ligated with pDA1836 digested with NotI and HindIII to remove the AmpR gene.
The lptL gene was amplified from pCB01 using primers P15 (5′-AACCATATGGAACCCGAGAACACCTTCAC-3′) and P16 (5′-CGCTCTAGAGTCAGGTCCACGCCGGCGAG-3′) and digested with NdeI and XbaI. The lptL gene was inserted into pDA2113 digested with NdeI and HindIII (removes SpcR and to terminator) as part of a three-piece ligation with the to terminator-CmR cassette, which was removed from pDA1842 as a HindIII and XbaI fragment, to produce pDA2117. The introduction of the to terminator-CmR cassette was required because the conjugation helper plasmid in ML22 is also NmR. The lptL expression plasmid was functionally validated by complementation of the DA1336 strain (ΔlptLI) to generate DA1470.
In a similar manner, pDA2129 was created by inserting the lptI gene amplified from pCB01, using primers P17 (5′-GAGGAATTCACATATGCACCGGCGGGAAC-3′) and P18 (5′-CAATCTAGACACTCCTCTGCGGGCAGTC-3′) and the Cm to terminator cassette digested with the appropriate restriction enzymes into pDA2113. The lptI expression plasmid was functionally validated by complementation of the DA613 ΔlptI mutant to generate DA1491.
Fermentation, measurement of lipopeptide production and purification of lipopeptides
Engineered strains of S. fradiae were fermented in DSF production medium,1 supplemented with 0.79% (w/v) of L-Ile (DSF-Ile).18 Addition of Ile to the optimized media drives production towards factors containing Ile13 and anteiso-undecanoyl side chains derived from precursors generated by Ile catabolism. Production of A54145 factors and novel lipopeptides was monitored by HPLC and LC-MS as described.18 Production of A54145 factors in liquid culture or on agar plates can also be assessed by a bioactivity assay using S. aureus ATCC 29213 with 5 mM CaCl2.18 Engineered strains producing novel lipopeptides were scaled up to 5 l of DSF-Ile in multiple shake flasks and centrifuged culture supernatant was purified by column chromatography using HP20 resin and semipreparative HPLC as described.24 Only the major lipopeptide produced in DSF-Ile media, containing Ile13 and anteiso-undecanoyl side chain was purified and characterized. LC-MS-MS analysis of select lipopeptides was carried out as described.24
Antibacterial activity assay for lipopeptides
Minimum inhibitory concentrations were determined by broth microdilution using Mueller–Hinton broth supplemented with 50 mg l−1 Ca2+ (MHBc). Cultures were incubated at 37°C with rotation (200 r.p.m.). Susceptibility of S. aureus ATCC 29213 for purified lipopeptides was determined in MHBc media with or without supplemented 1 % (v/v) bovine pulmonary surfactant (Survanta; Abbott Laboratories, Columbus, OH, USA).12
Bioinformatics analyses of putative Lpt proteins
BLASTP analysis25 of translated putative protein sequences was carried out at the NCBI website (http://www.ncbi.nlm.nih.gov/).
Results
Engineering and characterization of mutants blocked in the biosynthesis of hAsn3 and moAsp9, and assignment of lptJ, lptK and lptL gene functions
S. fradiae A54145 normally produces a mixture of lipopeptide factors containing Glu or 3mGlu at position 12, Ile or Val at position 13, coupled with any of three lipid side chains (Figure 1). In a previous study, it was shown that production of the factors containing 3mGlu12 can be eliminated by deleting the lptI methyltransferase gene in the biosynthetic gene cluster.18 Furthermore, factors A54145D and A54145E (containing Ile13 and with anteiso-undecanoyl side chains) can be greatly enriched by supplementing the fermentation medium with L-Ile. Bioinformatic analyses of the region downstream of the NRPS gene lptD (Figure 2) initially suggested that lptK, lptL and lptI genes are involved in amino acid modification, with the lptJ gene possibly involved in gene regulation based on an incorrect annotation of syrP (see below), and the lptM, lptN and lptP genes involved in resistance or transport.17 These analyses provided a strong rationale for the determination of lptJ, lptK and lptL gene functions by genetic modification, namely deletion and complementation.

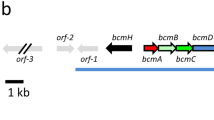
Genetic organization of a region of the lpt biosynthetic gene cluster, including the genes required for amino acid modification. Open reading frames are shown as arrows and the genes of interest are shown in gray and are located downstream from the NRPS (lptD) and NRPS-associated genes (lptG and lptH) in the gene cluster. The series of lines beneath the gene cluster show the extent of DNA present BACs or plasmids used in complementation experiments, with dotted lines representing S. fradiae DNA that continues beyond the region illustrated in the figure. For some of the plasmids gene expression is under the control of the ermE* promoter. lptI, methyltransferase involved in formation of 3mGlu12; lptJ, L-Asp9 hydroxylase; lptK, L-hAsp9 methyltransferase; lptL, L-Asn3 hydroxylase.
Using the tools and techniques developed for S. fradiae and described in Alexander et al.,18 a set of plasmids deleted for lptI, lptLI, lptKLI and lptJKLI was constructed by λ-Red-mediated recombination. The downstream ends of the lpt gene cluster containing BACs were deleted and replaced with the to terminator-AmpR cassette (Figure 2). The recombinant BACs were introduced into the lpt cluster deletion mutant DA1187 (ΔlptEF-I::tsr) and the engineered strains were fermented in DSF-Ile medium to characterize the lipopeptide(s) produced, with a focus only on the major lipopeptides produced containing Ile13 and anteiso-undecanoate side chains. Host strains, BACs, plasmids, their relevant genotype with respect to the lptJKLI genes for the engineered strains and the resulting lipopeptide products are summarized in Table 2.
A54145E, which contains hAsn3, moAsp9, 3mGlu12 and an anteiso-undecanoyl side chain, and has a mass ion of 1672.7, was used as the fully modified lipopeptide reference standard. Strain DA1243 (ΔlptI) produced A54145D, which has a mass ion of 1658.7 differing from A54145E by the loss of the Me group at Glu12 and consistent with previous work.18 Strain DA1336 (ΔlptLI) produced the major lipopeptide (CB-182,325), which had a mass ion of 1642.7, consistent with the loss of an additional hydroxyl group, indicating that lptL encodes one of the two hydroxylases needed. As this strain did not produce a compound lacking the methoxy group, lptL likely encodes the Asn3 hydroxylase. Strain DA1333 (ΔlptKLI) produced CB-182, 349, with a mass ion of 1628.7, consistent with the loss of an additional Me group. Therefore, lptK likely encodes the methyltransferase that converts hAsp9 to moAsp9. Strain DA1327 (ΔlptJKLI) produced CB-182,348 with a mass ion of 1612.7, consistent with the loss of an additional hydroxyl group, indicating that lptJ probably encodes the Asp9 hydroxylase.
Complementation of lpt mutants to generate novel lipopeptides
By introducing specific genes or gene sets into the mutants, it was possible to generate an expanded set of novel lipopeptides and further confirm the gene function assignments described above. Plasmid pDA2117 expressing the lptL gene was introduced into the DA1327 background giving DA1484, which produced CB-182,333, with a mass ion of 1628.7 (Table 2). When introduced into the DA1333 background, it generated DA1453, which produced CB-182,350, with a mass ion of 1644.7. The increase of 16 mass units to the parental lipopeptide is consistent with lptL encoding the Asn3 hydroxylase.
The plasmid pDA2129 expressing the lptI methyltransferase gene was introduced into the three deletion mutants described to give DA1489, DA1459 and DA1467, which produced three novel lipopeptides CB-182,390, CB-182,597 and CB-182,363, respectively. As expected, the mass of each of the three novel lipopeptides increased by 14 mass units from the parental lipopeptide consistent with its function as the Glu12 methyltransferase.18
Introduction of the pJR2015 BAC vector, containing the lptL and lptI genes, into DA1333 generated strain DA1627, which produced a novel lipopeptide with a mass ion of 1658.7, a 30 mass unit increase compared with the parental lipopeptide. Introduction of pJR2012 BAC vector with the lptK, lptL and lptI genes into DA1327 generated strain DA1553 and resulted in production of CB-182,443 with a 30 mass unit increase over the parental lipopeptide. An identical lipopeptide was produced when pJR2015 was introduced into DA1327, consistent with lptL encoding the Asn3 hydroxylase and lptI encoding the Glu12 methyltransferase, but suggests a lack of functional activity from the lptK methyltransferase present on the pJR2012 BAC on the hAsn3, Asp9 and 3mGlu12 lipopeptides produced by DA1553.
The engineered strains produced novel lipopeptides at titers that ranged from 32 to 360 mg ml−1 (Table 2). These titers were sufficient for compound isolation from the 5 l scale-up fermentations for further characterization.
LC-MS-MS characterization of select novel lipopeptides to confirm LptJ and LptL functions
Each of the engineered S. fradiae strains produced novel lipopeptides with mass ions consistent with the predicted gene functions for LptJ, LptK and LptL. Four of the compounds, A54145D (1), CB-182,333 (2), CB-182,348 (3) and CB-182,349 (4), were analyzed in detail to confirm the desired structural modifications. The structures of 1∼4 were supported by their HR-MS data at m/z 1658.7921 (Δ 0.43 p.p.m.), 1628.7790 (Δ 1.11 p.p.m.), 1612.7892 (Δ 2.05 p.p.m.) and 1628.7807 (Δ 0.06 p.p.m.) [M+H]+, respectively. Amino acid sequences of 1∼4 were determined by analysis of MS-MS data of both parent compounds (Figure 3a) and the linear hydrolysates 1a∼4a (Figure 3b). As summarized in Figure 3a, MS-MS spectra of 1∼4 provided limited but distinct product ions from y10 to y12 along with their corresponding water-loss peaks. The assigned three fragments y12, y11, y10 confirmed the side-chain amino acid sequences of 1 and 2 as anteiso-undecanoyl-Trp-Glu-hAsn and 3 and 4 as anteiso-undecanoyl-Trp-Glu-Asn, respectively. Furthermore, the amino acid sequences of 1∼4 were determined by the analyses of the MS-MS data from 1a∼4a (m/z 1676.8, 1646.8, 1630.7, 1646.8 [M+H]+, respectively), which were produced by hydrolysis of 1∼4 with lithium hydroxide. As shown in Figure 3b, experimental values of yn and bn agreed with their respective theoretical fragment ions. Therefore, the amino acid sequences of compound 1∼4 were strongly supported by the linear hydrolysates 1a-4a, as anteiso-undecanoyl-Trp-Glu-hAsn-Thr-Sar-Ala-Asp-Lys-moAsp-Gly-Asn-Glu-Ile, anteiso-undecanoyl-Trp-Glu-hAsn-Thr-Sar-Ala-Asp-Lys-Asp-Gly-Asn-Glu-Ile, anteiso-undecanoyl-Trp-Glu-Asn-Thr-Sar-Ala-Asp-Lys-Asp-Gly-Asn-Glu-Ile and anteiso-undecanoyl-Trp-Glu-Asn-Thr-Sar-Ala-Asp-Lys-hAsp-Gly-Asn-Glu-Ile, respectively. The MS-MS data are consistent with our gene function predictions such that CB-182,333 produced by DA1484 (ΔlptJKI), which expresses LptL, hydroxylates Asn3 and CB-182,349 produced by DA1333 (ΔlptKLI), which expresses LptJ, and hydroxylates Asp9.

(a) Chemical structures of compounds 1-4 and product ions from y10 to y12 of their LC-MS-MS spectra. (b) Chemical structures of linear hydrolysates 1a-4a with MS-MS fragmentation patterns and the corresponding product ions yn and bn.
Antibacterial properties of A54145 derivatives
The antibacterial properties of A54145 derivatives against S. aureus in the presence and absence of 1% bovine surfactant are shown in Table 3. As controls, daptomycin and its close analog CB-181,220, which has an anteiso-undecanoyl rather than an N-decanoyl side chain, were compared with the A54145 derivatives. Both daptomycin and CB-181,220 had MICs of 0.5 μg ml−1 in the absence of surfactant and 64 μg ml−1 in the presence of 1% surfactant. Potency of CB-182, 130, a Glu12 analog of CB-181,220 was significantly higher (16-fold higher MIC or 8 μg ml−1) than daptomycin or CB-181,220 in the absence of surfactant but retained activity in the presence of 1% surfactant (16 μg ml−1). A54145E had an MIC of 1 μg ml−1 in the absence of surfactant, but the MIC in the presence of 1% surfactant was increased by 32-fold. Removal of the Me group from Glu12 (A54145D) resulted in only a twofold increase in MIC (2 μg ml−1) in the absence of surfactant, and an eightfold reduction in MIC in the presence of surfactant (4 μg ml−1) relative to A54145E. A similar result was obtained by removing the methoxy group from Asp9 (CB-182,443). Removal of the methoxy group from Asp9 and the hydroxyl group at Asn3 yielded CB-182,390, which had MICs of 2 μg ml−1 in the presence or absence surfactant. Other combinations of hypo-modification of Asn3, Asp9 and Glu12 gave antibiotics with less favorable MIC profiles (Table 3).
The results from inactivating the various amino acid modification enzymes indicate that the antibacterial properties of A54145 can be altered by simply changing the modified amino acid composition. Whereas the fully modified A54145E was the most active antibiotic, several compounds lacking one or two amino acid modifications were nearly as active as A54145E in the absence of surfactant and more active in the presence of surfactant. The best compound (CB-182,390) displayed only a twofold in increase in MIC (2 μg ml−1) over A54145E in the absence of surfactant, with no change in potency in the presence of surfactant (2 μg ml−1).
Bioinformatic analysis of the lptJKL gene products
LptL showed highest sequence similarity (47%) to AsnO that hydroxylates Asn during the lipopeptide CDA biosynthesis.26, 27 Furthermore, LptL has a binding pocket for the oxygenation of Asn identical to that of AsnO, EQNHENDHR (Table 4).27 LptL and AsnO homologs with amino acid sequences ranging from 47 to 60% identities were observed in Saccharopolyspora erythraea, Streptomyces scabies, Streptomyces sp. SPB78, Streptomyces sp. SPB74 and Streptomyces sp. C, all of which have amino acid binding pockets identical to those of AsnO and LptL (Table 4). S. coelicolor and Streptomyces lividans encode AsnO proteins of identical sequence, and also encode AsnO/LptL homologs with binding pockets differing in one amino acid from the AsnO/LptL pocket (Table 4).
Although LptJ encodes a hydroxylase that converts Asp9 to hAsp9, BLASTP analysis indicated that it has no sequence similarity to LptL. The top BLASTP hits were to a conserved hypothetical protein from Streptomyces clavuligerus (ZP_050077403; 63% identity) and a SyrP-like protein from Streptomyces avermitilis (NP_824815; 63% identity). It also showed 33% identity with the SyrP L-Asp hydroxylase from Pseudomonas syringae.28 LptJ defines the first L-Asp hydroxylase encoded by a secondary metabolite biosynthetic gene cluster in an actinomycete, although it has been shown that asnO can be modified by site-specific mutagenesis to encode an enzyme that will hydroxylate Asp.29
BLASTP analysis of LptK was consistent with it functioning as a methyltransferase. However, the only BLASTP hit above 31% identity was to a putative methyltransferase from Streptomyces griseus (BAG17399; 45% sequence identity).
Discussion
The cyclic lipopeptide antibiotic A54145 complex of related factors is distantly related to the clinically important antibiotic daptomycin, and several of the factors demonstrate good antibacterial activities against Gram-positive pathogens.1, 8 The most active compounds containing 3mGlu12 were toxic to mice, whereas those containing Glu12 were about 20-fold less toxic and displayed only twofold lower antibacterial activities.8 In a recent study, we generated strains deleted for the lptI gene, and the mutants produced >0.5 g l−1 of the Glu12-containing A54145D when fermentations were supplemented with L-Ile.18
In the present study, we generated a series of S. fradiae strains deleted for different combinations of the lptJ, lptK, lptL and lptI genes. By analyzing the products of the engineered strains, we were able to deduce the functions of the lptJ, lptK and lptL genes, as Asp9 hydroxylase, hAsp9 methyltransferase and Asn3 hydroxylase, respectively, and further confirmed the function of the lptI gene as the Glu12 methyltransferase. The genetic engineering tools developed for S. fradiae were well suited for a combinatorial biosynthesis approach to assign gene function and generate a series of novel lipopeptides. Fermentation of the engineered strains in DSF-Ile media shifted production predominantly to the factors containing Ile13 and anteiso-undecanoyl side chains, which simplified HPLC and LC-MS analysis of the fermentation broths and purification of the novel A54145 factors. We chose to pursue the purification and characterization of only A54145D or A54145E analogs (Ile13 and the anteiso-undecanoyl side chain) because of the concentration advantage afforded by the Ile supplementation and increased chromatographic separation of these lipopeptides resulting in higher-yielding purifications. Characterization of novel lipopeptides with the different lipid side chains would have increased the number of analogs generated, but the MIC data8 did not suggest significant differences among the different side chains to warrant purification of the remaining lipopeptides. Also in support of this decision to focus only on the major lipopeptide, we had confirmed the previous observation that Val13-containing lipopeptides A54145F had inferior MICs in comparison with the Ile13 counterpart (Counter et al.8; and data not shown). With all analogs having the same lipid side chain, direct antibacterial activity comparison among the hybrid lipopeptides was possible.
It is noteworthy that even the strain deleted for all four genes produced lipopeptide antibiotics (CB-182,348; Table 2). This means that the NRPS enzymes can process Asn or hAsn at position 3, Asp, hAsp or moAsp at position 9 and Glu or 3mGlu at position 12. This astonishing flexibility was exploited to generate many novel compounds, discussed below. It is also noteworthy that the specific amino acid modifications were localized to the L-isomers of Asn, Asp and Glu at positions 3, 9 and 12, respectively, even though A54145E has a D-Asn, L-Asp and D-Glu residues at positions 11, 7 and 2, respectively. This site specificity of amino acid modifications is particularly relevant in light of the observation that the amino acid binding pockets for hAsn3 and moAsp9 are indistinguishable from those for Asn11 and Asp7, respectively.17 Further characterization of AsnO and its functional homolog LptL and their interactions with their respective NRPS proteins may shed light on the mechanism of relaxed amino acid binding specificity at positions 3, 9 and 12 for modified amino acids, coupled with the apparent stringent amino acid binding specificity at positions 2, 7 and 11.
Addition of pJR2012 (lptKLI) or pJR2015 (lptLI) to DA1327 (ΔlptJKLI) generated strains that produced the hAsn3, Asp9 and 3mGlu12 lipopeptide CB-182,443. It is not a lack of function from the lptK methyltransferase present on the pJR2012, but rather lptK function requires the presence of lptJ to generate the hAsp9 substrate necessary for lptK O-methylation. This sequential order was confirmed in an additional strain engineering experiment using plasmids expressing only lptJ or only lptK or lptJ and lptK together in the DA1327 background (data not shown). The strain containing the lptK plasmid alone produced only the parental lipopeptide, whereas lptJ alone and lptJK together produced lipopeptides with 16 and 30 mass unit increases to the parental lipopeptide, confirming lptK action on the lptJ catalyzed hAsp9 substrate. These data further confirm that LptJ converts Asp9 to hAsp9, the obligatory substrate for the LptK O-methyltransferase action.
Titers of the novel lipopeptides ranged from 10 to 75% of the 462 mg ml−1 of A54145D produced by DA1243. The majority of the novel lipopeptide producing strains lacked the LptM and LptN transporter pair, which are clearly not essential for antibiotic export, but may aid in optimal export of antibiotic. The lptM and lptN genes are only present in strains DA1243, DA1553 and DA1627. Strain DA1453 produces the hAsn3, hAsp9, Glu12 (CB-182,350) at ∼75% of the A54145D titers, whereas hAsn3, Asp9, Glu12 (CB-182,333) is produced at only ∼25% by strain DA1484 and Asn3, moAsp9, Glu12 (CB-182,325) is produced at only ∼17% by strain DA1336. This type of direct comparison would be more conclusive with individual in-frame deletion mutants for each gene, but it does suggest less efficient production of Asn3 or Asp9-containing lipopeptides. Fermentation of S. fradiae XH25 typically generates a mixture of approximately 60% Glu12 and 40% 3mGlu12 factors, suggesting a shortage of 3mGlu or precursors or that there is an incorporation or processing preference for the Glu12-containing peptides. Interestingly, in most cases the novel 3mGlu12-containing lipopeptides generated in this study represented >75% over their Glu12-containing counterpart produced during the fermentation.
MIC data demonstrated that modified amino acids are important for activity; this was especially true for the Glu12 analogs. In all cases, the 3mGlu12-containing analogs are more active than their Glu12 counterparts. Similar to A54145D, novel Glu12-containing lipopeptides displayed substantially lower MICs in the presence of bovine surfactant, but their potency is not sufficient for clinical consideration. It is noteworthy that the analogs CB-182,443 and CB-182,390 have 3mGlu12, which results in good potency but uniquely displayed good activity in the presence of 1% surfactant. CB-182,390 (Asn3, Asp9 and 3mGlu12) was targeted for further investigation, including extensive chemical analyses, including amino acid quantitation, tandem LC-MS-MS analysis and 2D-NMR to confirm the structure.24
Potent antibacterial activity depends on the presence of modified amino acids, making total peptide synthesis or chemoenzymatic synthesis,30 utilizing proteinogenic amino acids undesirable approaches to generate A54145 analogs. As chemical synthesis of the chiral modified amino acids at the necessary scale for SAR studies is not practical, combinatorial biosynthesis19 remains the most effective method to generate potent novel lipopeptides.
In a separate study, the M domain was deleted from the Sar5 module to generate an A54145D analog (CB-182,391) containing Gly5, which was eightfold less active than A54145D in the presence or absence of surfactant with MICs of 16 and 32 μg ml−1, respectively (S Doekel and P Brian, unpublished). This indicates that Sar5 is also important for overall activity of A54145 lipopeptides.
The study by Strieker et al.27 have shown that AsnO converts the free L-Asn to L-hAsn before incorporation into the growing peptide. According to their model, L-hAsn binds to the adenylation (A)-domain of the CAhAsn9TE module of CdaPS2, in which it is converted to D-hAsn and incorporated into the growing peptide chain. The AsnO protein was crystallized, and the catalytic binding pocket for L-Asn has been defined as a 10 letter code: EQNHENDHR at positions 125, 144, 146, 155, 157, 158, 241, 287 and 305.27 The LptL protein has an identical L-Asn catalytic binding pocket displaced by eight amino acids relative to AsnO, and starting at position E117. Therefore, even though the lptL and asnO genes and their protein products have diverged considerably (47% identical amino acids), they have conserved the L-Asn binding pocket 100%. This supports the notion that although these enzymes have orthologous catalytic functions, they have likely diverged for a second non-orthologous function.31 One possible non-orthologous function is binding to different NRPS enzymes at specific modules to provide L-hAsn in situ on demand during peptide assembly.31 This model predicts amino acid sequence divergence in regions of the proteins involved in protein-protein interactions, and also provides a mechanism to sequester L-hAsn from the cytoplasmic amino acid pools that can be drawn on for protein biosynthesis. It also explains the absence of incorporation of hAsn at position 11.
There are currently over 20 streptomycete and many other actinomycete genome sequences in GenBank. There are homologs to LptL and AsnO showing >47% amino acid identities over nearly the entire proteins in seven streptomyctes and in Saccharopolyspora erythraea (Table 4). Their high amino acid sequence similarities suggest that some or all may function as L-Asn oxygenases.
BLASTP analysis indicated that LptJ is unrelated to the LptL, but it displays high sequence similarities with only two proteins, a SyrP-like protein from S. avermitilis (62% identity) and a conserved hypothetical protein from S. clavuligerus (62% identity). LptJ shows 33% sequence identity with SyrP from P. syringae, an enzyme that has been shown to hydroxylate L-Asp tethered as the protein-bound S-pantetheinyl thioester on an NRPS module involved in syringomycin biosynthesis to produce L-threo-3-OH-Asp.28 If LptJ hydroxylates L-Asp in a similar manner during A54145 biosynthesis, it might account for the specificity of hydroxylation at Asp9 and not Asp7. LptJ defines functionally the first of the SyrP-like L-Asp hydroxylases in actinomycetes, and the high sequence identities to the two related proteins from genome sequencing projects suggests that they may also function as L-Asp hydroxylases. Bioinformatics had originally predicted LptJ to have a role in gene regulation based on its initial annotation.15 Biochemical characterization of SyrP confirmed its actual role in syringomycin biosynthesis is as an Asp hydroxylase,28 which is consistent with our prediction for the homologous LptJ in this study.
BLASTP analysis of LptK indicated that this protein class is even more obscure that LptJ. LptK showed 45% sequence identity to a putative methyltransferase from S. clavuligerus and 45% identity to a sequence (likely a methyltransferase) embedded in a NRPS from Pseudomonas aeruginosa strains. The uniqueness of LptK may not be surprising as the moAsp in A54145 represents the only known moAsp-containing natural product that we are aware of. The functional assignment of the lpt genes described herein should help in the annotation of cryptic secondary metabolite pathways that are being rapidly discovered by genome mining.
References
Boeck, L. D. et al. A54145, a new lipopeptide antibiotic complex: discovery, taxonomy, fermentation and HPLC. J. Antibiot. 43, 587–593 (1990).
Fukuda, D. S., Du Bus, R. H., Baker, P. J., Berry, D. M. & Mynderse, J. S. A54145, a new lipopeptide antibiotic complex: isolation and characterization. J. Antibiot. 43, 594–600 (1990).
Debono, M. et al. A21978C, a complex of new acidic peptide antibiotics: isolation, chemistry, and mass spectral structure elucidation. J. Antibiot. 40, 761–777 (1987).
Debono, M. et al. Enzymatic and chemical modifications of lipopeptide antibiotic A21978C: the synthesis and evaluation of daptomycin (LY146032). J. Antibiot. 41, 1093–1105 (1988).
Boeck, L. D. & Wetzel, R. W. A54145, a new lipopeptide antibiotic complex: factor control through precursor directed biosynthesis. J. Antibiot. 43, 607–615 (1990).
Boeck, L. D., Fukuda, D. S., Abbott, B. J. & Debono, M. Deacylation of A21978C, and acidic lipopeptide antibiotic complex, by Actinoplanes utahensis. J. Antibiot. 41, 1085–1092 (1988).
Fukuda, D. S., Debono, M., Molloy, R. M. & Mynderse, J. S. A54145, a new lipopeptide antibiotic complex: microbial and chemical modification. J. Antibiot. 43, 601–606 (1990).
Counter, F. T. et al. A54145, a new lipopeptide antibiotic complex: microbiological evaluation. . J. Antibiot. 43, 616–622 (1990).
Arbiet, R. D., Maki, D., Tally, F. P., Campanaro, E. & Eisenstein, B. I. The safety and efficacy of daptomycin for the treatment of complicated skin and skin-structure infections. Clin. Infect. Dis. 38, 1673–1681 (2004).
Fowler, V. G. et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N. Engl. J. Med. 355, 653–655 (2006).
Pertel, P. E. et al. Effects of prior effective therapy on the efficacy of daptomycin and ceftriaxone for the treatment of community-acquired pneumonia. Clin. Infect. Dis. 46, 1142–1151 (2008).
Silverman, J. A., Morton, L. I., Vanpraagh, A. D., Li, T. & Alder, J. Inhibition of daptomycin by pulmonary surfactant: in vitro modeling and clinical impact. J. Infect. Dis. 191, 2149–2152 (2005).
Hill, J. J. et al. Synthesis and biological activity of N-acylated ornithine analogues of daptomycin. Bioorg. Med. Chem. Lett. 13, 4187–4191 (2003).
Siedlecki, J et al. Array synthesis of novel lipodepsipeptide. Bioorg. Med. Chem. Lett. 13, 4245–4249 (2003).
Miao, V. et al. Genetic engineering in Streptomyces roseosporus to produce hybrid lipopeptide antibiotics. Chem. Biol. 13, 269–276 (2006).
Nguyen, K. T. et al. Combinatorial biosynthesis of lipopeptide antibiotics related to daptomycin. Proc. Natl Acad. Sci. USA 103, 17462–17467 (2006).
Miao, V., Brost, R., Chapple, J., Coëffet-LeGal, M.- F. & Baltz, R. H. The lipopeptide antibiotic A54145 biosynthetic gene cluster from Streptomyces fradiae. J. Ind. Microbiol. Biotechnol. 33, 129–140 (2006).
Alexander, D. C. et al. Development of a genetic system for lipopeptide combinatorial biosynthesis in Streptomyces fradiae and heterologous expression of the A54145 biosynthetic gene cluster. Appl. Environ. Microbiol. 76, 6877–6887 (2010).
Nguyen, K. T. et al. Genetically engineered lipopeptide antibiotics related to A54145 and daptomycin with improved properties. Antimicrob. Agents Chemother. 54, 1404–1413 (2010).
Mahlert, C., Kopp, F., Thirlway, J., Micklefield, J. & Marahiel, M. A. Stereospecific enzymatic transformation of α-ketoglutarate to (2S,3R)-3-methyl glutamate during acidic lipopeptide biosynthesis. J. Am. Chem. Soc. 129, 12011–12018 (2007).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: a Laboratory Manual 2nd edn. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989).
Datsenko, K. A. & Wanner, B. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA 97, 6640–6645 (2000).
Gregory, M. A., Till, R. & Smith, M. C. M. Integration site for Streptomyces phage φBT1 and development of site-specific integrating vectors. J. Bacteriol. 185, 5320–5323 (2003).
Gu, J.-Q. et al. Structural characterization of a lipopeptide antibiotic A54145E (Asn3Asp9) produced by a genetically engineered strain of Streptomyces fradiae. J. Antibiot (e-pub ahead of print 24 November 2010; doi:10.1038/ja.2010.140).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Hojati, Z. C. et al. Structure, biosynthetic origin, and engineered biosynthesis of calcium-dependent antibiotics from Streptomyces coelicolor. Chem. Biol. 9, 1175–1187 (2002).
Streiker, M., Kopp, F., Mahlert, C., Essen, L.- O. & Marahiel, M. A. Mechanistic and structural basis of stereospecific Cβ-hydroxylation in calcium-dependent antibiotic, a daptomycin-type lipopeptide. ACS Chem. Biol. 2, 187–196 (2007).
Singh, G. M., Fortin, P. D., Koglin, A. & Walsh, C. T. Beta-hydroxylation of the aspartyl residue in the phytotoxin syringomycin E: characterization of two candidate hydroxylases AspH and SyrP in Pseudomonas syringae. Biochemistry. 47, 113010–113020 (2008).
Streiker, M., Essen, L.- O., Walsh, C. T. & Marahiel, M. A. Non-heme hydroxylase engineering for simple enzymatic synthesis of L-threo-hydroxyaspartic acid. Chem. Bio. Chem. 9, 374–376 (2008).
Kopp, F., Grunewald, J., Mahlert, C. & Marahiel, M. A. Chemoenzymatic design of acidic lipopeptide hybrids: new insights into the structure-activity relationship of daptomycin and A54145. Biochemistry 45, 10474–10481 (2006).
Baltz, R. H. Biosynthesis and genetic engineering of lipopeptide antibiotics related to daptomycin. Curr. Top. Med. Chem. 8, 618–638 (2008).
Acknowledgements
We thank Maggie Smith of the University of Aberdeen for providing plasmid pRT802. We are grateful to members in the Analytical Chemistry, Natural Products Chemistry and Natural Products Fermentation departments at Cubist for technical assistance. This work was supported by Cubist Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to late Dr C Richard ‘Dick’/’Hutch’ Hutchinson for his exceptional contributions to natural product biosynthesis, engineering and drug discovery.
Rights and permissions
About this article
Cite this article
Alexander, D., Rock, J., Gu, JQ. et al. Production of novel lipopeptide antibiotics related to A54145 by Streptomyces fradiae mutants blocked in biosynthesis of modified amino acids and assignment of lptJ, lptK and lptL gene functions. J Antibiot 64, 79–87 (2011). https://doi.org/10.1038/ja.2010.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.138
Keywords
This article is cited by
-
Synthetic biology, genome mining, and combinatorial biosynthesis of NRPS-derived antibiotics: a perspective
Journal of Industrial Microbiology and Biotechnology (2018)
-
Translocation of the thioesterase domain for the redesign of plipastatin synthetase
Scientific Reports (2016)
-
Harnessing natural product assembly lines: structure, promiscuity, and engineering
Journal of Industrial Microbiology and Biotechnology (2016)
-
Natural product discovery: past, present, and future
Journal of Industrial Microbiology and Biotechnology (2016)
-
Therapeutic cyclic lipopeptides mining from microbes: latest strides and hurdles
World Journal of Microbiology and Biotechnology (2015)
