
Abstract
Our current knowledge of plant-microbe interactions indicate that populations inhabiting a host plant are not restricted to a single microbial species but comprise several genera and species. No one knows if communities inside plants interact, and it has been speculated that beneficial effects are the result of their combined activities. During an ecological study of nitrogen-fixing bacterial communities from Lupinus angustifolius collected in Spain, significant numbers of orange-pigmented actinomycete colonies were isolated from surface-sterilized root nodules. The isolates were analysed by BOX-PCR fingerprinting revealing an unexpectedly high genetic variation. Selected strains were chosen for 16S rRNA gene sequencing and phylogenetic analyses confirmed that all strains isolated belonged to the genus Micromonospora and that some of them may represent new species. To determine the possibility that the isolates fixed atmospheric nitrogen, chosen strains were grown in nitrogen-free media, obtaining in some cases, significant growth when compared with the controls. These strains were further screened for the presence of the nifH gene encoding dinitrogenase reductase, a key enzyme in nitrogen fixation. The partial nifH-like gene sequences obtained showed a 99% similarity with the sequence of the nifH gene from Frankia alni ACN14a, an actinobacterium that induces nodulation and fixes nitrogen in symbiosis with Alnus. In addition, in situ hybridization was performed to determine if these microorganisms inhabit the inside of the nodules. This study strongly suggests that Micromonospora populations are natural inhabitants of nitrogen-fixing root nodules.
Similar content being viewed by others

Introduction
Plants are constantly involved in interactions with a wide range of soil bacteria which colonize the rhizosphere, the phyllosphere and inside of plant tissues (Zakhia et al., 2006). Rhizobia are soil microorganisms that can live symbiotically in the roots of leguminous plants and have a key role in nitrogen fixation. These α- and β-proteobacteria are capable of inducing nodules on the roots of their plant hosts and reduce dinitrogen. Endophytic nitrogen-fixing bacteria seem to constitute only a small proportion of total endophytic bacteria (Rosenblueth and Martinez-Romero, 2006). Our current knowledge of plant–microbe interactions indicate that populations inhabiting a host plant are not restricted to a single microbial species but comprise several genera and species.
In recent studies, Coombs and Franco (2003) reported on the isolation of endophytic actinobacteria from healthy wheat tissues and showed their potential use as biological control agents (Coombs et al., 2004). Furthermore, Tokala et al. (2002) showed that a Streptomyces strain, WYEC108, was able to colonize the roots of Pisum sativum increasing root nodulation frequency. These authors are of the opinion that these plant–microbe interactions may be common in nature.
The genus Micromonospora belongs to the phylum Actinobacteria. Typically, colonies on agar media are initially light orange, becoming orange, red, brown or purple with the production of spores. Micromonospora is the type genus of the family Micromonosporaceae and contains microorganisms with potential biotechnological applications (Ismet et al., 2004; Thawai et al., 2004). Micromonosporae have been isolated from diverse habitats including soil, water and marine sediments (Kawamoto, 1989; Zhao et al., 2004; Maldonado et al., 2005; de Menezes et al., 2008). Recently, strains of this genus have also been recovered from diverse plant tissues, especially nitrogen-fixing nodules (Valdés et al., 2005; Trujillo et al., 2006, 2007; Garcia et al., 2010). In the last 4 years our research group has isolated many Micromonospora strains from nitrogen-fixing nodules of different legume species. To date, we maintain a collection of more than 500 Micromonospora isolates obtained from legume plants collected in Spain, Ecuador and Nicaragua. Thus, we have isolated strains from plants of Arachis hypogaea, Cicer arietinum, Glycine max, Lens culinarium, Lupinus angustifolius, Lupinus gredensis, Medicago sativa, Ononis sp., Ornithopus sp., P. sativum, Trifolium sp., Vicia sp. and the tropical legume Mucuna sp. Indeed, we have found Micromonospora strains in the root nodules of every legume sampled so far (Trujillo et al., 2007; Cerda, 2008; Garcia et al., 2010).
Frankia is a nitrogen-fixing actinobacterium that can grow as a microsymbiont in the root nodules of several woody dicotyledonous plants. Similarly to rhizobia, Frankia strains induce root nodules on their host plants and supply them with atmospheric nitrogen. The report of two isolates from Casuarina nodules identified as members of the families Thermomonosporaceae and Micromonosporaceae that contained nifH genes with a sequence similar to those of Frankia, strongly suggests that Micromonospora may also possess the ability to fix nitrogen (Valdés et al., 2005).
In this study we describe the isolation, diversity and phylogenetic relationship of Micromonospora populations isolated from the root nodules of naturally occurring L. angustifolius plants collected in various geographical regions in Spain. In addition, we studied the capacity of some of these strains to grow in nitrogen-free media and determined the presence of nifH-like sequences in their genomes. In addition, we used fluorescent in situ hybridization (FISH) to detect the presence of Micromonospora inside the root nodules. Finally, a 500 bp DNA fragment corresponding to the gyrase B gene of Micromonopsora was detected by direct amplification of DNA isolated from nodules. These findings suggest that Micromonospora is part of the indigenous bacterial community inhabiting legume nitrogen-fixing nodules, which is still poorly understood.
Materials and methods
Plant sampling and soil characterization
Naturally occurring plants of L. angustifolius were collected from the regions of Castilla y León (Cabrerizos, 40°58′43″ N–5° 36′ 46″ W, and Saelices 40°40′06″ N–6° 38′ 02″ W) and Extremadura (Plasencia, 39°58′38″ N–6°2′26″ W) located in the mid and southwest Spain, respectively, and characterized by a Mediterranean climate. Different types of soils have developed from the parent materials of the area. There are acid soils on granites (Saelices) and slates (Plasencia) whereas basic soils over sandstones and conglomerates are found in Cabrerizos. The soil attached to the roots (rhizhosphere and rhizoplane) was used to determine various physico-chemical parameters. Soil samples were air-dried and screened to 2 mm and analyses were performed with the fine-earth fraction (<2 mm). The pH was measured potentiometrically in a 1:1 soil-water suspension, in a CRISON digit, micropH 2001 instrument. Organic matter was determined by potassium-dichromate oxidation as described by Walkley (1947) and total organic N analyses were conducted by the Kjeldahl method (Bremner and Mulvaney, 1982). Available Ca, Mg and K were extracted with 1M ammonium acetate and determined by plasma ICP-OES ULTIMA-2 Jovin-Yvon mod. In addition, available P was determined by the Bray II method, modified from Bray and Kurtz (1945).
Isolation of microorganisms
Four to five plants were collected from each site for three consecutive years; sterile pots were used to transfer the samples to the laboratory for immediate isolation of the microorganisms. Root systems were thoroughly washed with sterile distilled water to remove attached soil, followed by sonication to dislodge any possibly trapped soil pockets. Clean root nodules were either used for bacterial isolation or stored in glycerol (20% v v−1) at −80 °C for in situ detection studies. Nodules for isolation of microorganisms (2–4) were selected mainly on the basis that they were effective nodules, that is, a pink–red colour indicating the presence of nitrogen fixation activity. Isolations were made according to the method of Vincent (1970) for the recovery of nitrogen-fixing bacteria. Briefly, the washed nodules were surface sterilized with HgCl2 (2.5%, w/v) for 2 min, rinsed five times with sterile distilled water and aseptically crushed using a sterile glass rod. The macerate was inoculated on yeast mannitol agar (YMA, Vincent, 1970) under aseptic conditions and the plates were incubated for 3 weeks at 28 °C. No antibiotics or antifungals were added to the isolation medium. For each isolation experiment a sterile, non-crushed nodule was rolled over YMA agar and incubated under the same conditions as the homogenized samples. A washed non-sterile nodule was also included as control. Sixty-four nodules were processed: 29 originated from nine plants collected in Cabrerizos; 32 nodules from 12 plants collected in Plasencia and 10 nodules from four plants collected in Saelices. Colonies on isolation plates were selected on the basis of their morphological appearance as described for Micromonospora (Kawamoto, 1989) and picked under a stereoscopic microscope, checked for purity and streaked on YMA to obtain pure cultures. In addition, the strains were Gram-stained using the procedure of Doetsch (1981).
DNA isolation
All bacterial strains isolated were grown on International Streptomyces Project 2 agar (Shirling and Gottlieb, 1966) for 5 days at 28 °C. A single colony from each culture was washed in Tris-EDTA (50 mM Tris-HCl pH 8.0, 10 mM EDTA) followed by centrifugation at 10 000 g for 5 min. DNA extraction was performed using the REDExtract-N-Amp Plant PCR kit (Sigma, Steinheim, Germany) following the manufacturer's instructions but with an additional purification step using phenol/chloroform.
BOX–PCR profiling
BOX–PCR fingerprinting profiles from bacterial genomic DNA were generated using the BOXA1R primer for the 136 isolates (Versalovic et al., 1994). PCR amplifications for each strain were performed using the PCR mixture included in the REDExtract-N-Amp Plant PCR kit (Sigma) in a final volume of 20 μl per reaction and following the manufacturer's recommendations. Thermal cycling parameters were as follows: 7 min at 95 °C, 30 cycles of 1 min at 94 °C, 1 min at 52 °C and 3 min at 72 °C, followed by 10 min final extension at 72 °C. Ten microliters of each PCR product were loaded on a 2% agarose gel (15 cm × 20 cm) containing 0.5 μg ml–1 ethidium bromide. Electrophoresis was run at 75 V for 3 h in freshly prepared 1X TBE-EDTA buffer at pH 8.0 using a Bio-Rad powerPac 300 power supply. DNA molecular weight marker XIV (Roche, Mannheim, Germany) was used as a molecular size standard. After electrophoresis gels were photographed, stored on disk as TIFF files and imported into the software package BioNumerics version 4.5 (Applied Maths, Sint-Martens-Latem, Belgium). Similarity matrices of densitometric curves of the gel tracks were calculated using the Pearson Product Moment Correlation Coefficient followed by tree construction using the UPGMA algorithm. Strain clusters were defined at the 60% level of similarity.
Multi-dimensional scaling analysis
All BOX–PCR fingerprints were also compared by scoring similarities using the Pearson's moment coefficient and then building a multidimensional scaling map from the resulting similarity matrix to obtain information about the relationships among the microorganisms isolated from the three different locations.
Amplification and sequencing of the 16S rRNA gene
The 16S rRNA gene of 64 strains selected from the 37 BOX clusters (1–3 strains for cluster) were amplified using the REDExtract-N-Amp Plant PCR kit (Sigma) in 20 μl reactions using the universal primer set 27f and 1522r (Lane, 1991). Amplification and sequencing conditions were as follows: 9 min at 94 °C, 35 cycles of 1 min at 94 °C, 1 min at 56 °C and 2 min at 72 °C, followed by 7 min final extension at 72 °C. PCR products were electrophoresed in 1% agarose gels containing ethidium bromide (0.5 μg ml−1) using modified Tris-acetate EDTA buffer (Millipore, Cork, Ireland). The amplified bands were excised and purified using the QIAquick gel extraction kit (Qiagen, Hilden, Germany) according to the manufacturers’ instructions. The sequence reactions were performed on an ABI377 sequencer (Applied Biosystems, Foster City, CA, USA) using a BigDye terminator v3.0 cycle sequencing kit as supplied by the manufacturer and using the primers described previously (Rivas et al., 2003). The sequences were manually aligned using clustal X (Thompson et al., 1997) and compared with other sequences deposited in public databases (GenBank/EMBL) and the Eztaxon server (http://www.eztaxon.org/; Chun et al., 2007). Phylogenetic distances were calculated with the Kimura 2-parameter model (Kimura, 1980) and the tree topologies were inferred using the neighbour-joining (Saitou and Nei, 1987) method using 1000 bootstrap replications. All analyses were carried out using the MEGA 4 program (Tamura et al., 2007).
Growth in nitrogen-free media
Thirty-seven strains (one representative from each cluster) were grown in nitrogen-free semisolid agar to determine their potential nitrogen-fixing activity. Bradyrhizobium sp. ISLU 65 (Jarabo-Lorenzo et al., 2003) was used as a strain control. Strains grown on ISP 2 agar were point inoculated in the bottom of test tubes with 10 ml semi-solid agar, which contained 1% Yeast Carbon Base (Difco, Sparks, MD, USA) and 1% Noble Agar (Difco). The strains were incubated in the dark at 28 °C for 3 weeks. After the first incubation period, the strains were transferred to fresh media using the first tubes as inoculating source and incubated for an additional 3-week period. The above medium supplemented with (NH4)2SO4 (2 g l−1) was used as a positive control.
Amplification and sequencing of the nifH gene
A combination of touchdown and nested PCR was used to screen for the presence of nifH genes in the strains that grew in nitrogen-free media. Different protocols were assayed to successfully amplify the nifH from 7 strains. In the case of isolate Lupac 08, a nifH-like fragment was amplified and sequenced using the primers and procedure described by Valdés et al. (2005). The remaining six isolates were amplified using a two-step PCR. The first fragment of ∼650 bp was amplified from genomic DNA using the REDExtract-N-Amp Plant PCR kit mixture (Sigma) as described before, the primers used were MGf (5′-CACGGATCCGCAAGGGTGGTATT-3′) and MGr2 (5′-TGAAGCTTCTCGATGACCGTCATCCG-3′) (Normand et al., 1988). Thermal cycler conditions for touchdown PCR program were: initial step of 3 min at 95 °C, 10 cycles of 25 s at 94 °C, 30 s at 64 °C and 40 s at 72 °C, with a 0.4 °C decrease of the annealing temperature every cycle. After completion of the touchdown program, 30 additional cycles were performed under the following conditions: 25 s at 94 °C, 30 s at 57 °C and 40 s at 72 °C ending with a final extension of 5 min at 72 °C.
The second (nested) PCR amplified an internal region of ∼350-bp fragment using the primers ELA1 (5′-ATGGCKGCCATGGCCGAG-3′) (Gtari et al., 2007) and PolR (5′-ATSGCCATCATYTCRCCGGA-3′) (Poly et al., 2001). The template for the second PCR was 1 μl of the touchdown PCR product. Reactions were carried out in 50 μl reactions, each containing 5 μl 10 × buffer (Applied Biosystems), 4 μl 25 mM MgCl2 (Applied Biosystems), 2 μl 0.1% seroalbumin (Sigma), 1.25 μl dNTPs (10 mM each, Applied Biosystems), 0.5 μl each primer (20 μM), 0.4 μl (5U/μl) Taq Gold DNA polymerase. PCR conditions were: 9 min at 95 °C, 30 cycles of 1 min at 94 °C, 1 min at 57 °C, 2 min at 72 °C and a final extension of 7 min at 72 °C. PCR product purification and sequencing were performed as described above using the PolR primer. A third PCR product was obtained for strain Lupac 09 using primers ELA1 and MGr2 under the following conditions: 9 min at 95 °C, 30 cycles of 1 min at 94 °C, 1 min at 64 °C and 2 min at 72 °C ending with a final extension of 7 min at 72 °C. This fragment was subsequently sequenced using primer NF462 (5′-AGGCCGGCGCGTACGAGGAC-3′) designed in this study. The sequenced fragment, which overlapped with the 350 pb obtained before was used to construct a single contig of 619 bp.
Design and labelling of probes for FISH
Two probes complementary to the 16S rRNA gene of Micromonospora and Bradyrhizobium were designed after sequence alignment of the 16S rRNA gene sequences of available Micromonospora and Bradyrhizobium type strains and the sequences obtained in this study. The probes MicromCy5.5 (5′-CTGGAGGTTTTGCGGCCA-3′, E. coli position 1000) and IsluCy5.5 (5′-AGGGTTGCGCCCATTGTCC-3′, E. coli position 369) were checked for possible targets against the Genbank (Blast) and the ribosomal database project II databases (Probe Match program; http://rdp.cme.msu.edu/probematch/search.jsp). In both cases only hits identified as Micromonospora or Bradyrhizobium sequences were obtained. The oligonucleotides were labelled with the fluorochrome Cy5.5 (indodicarbocyanine) and were synthesized by Isogen (De Meern, Netherlands). The universal probe EU338 5′-GCTGCCTCCCGTAGGAGT-3′ (Amann et al., 1995) labelled with FITC was used to optimize permeabilization and hybridization parameters.
Pretreatment and sample fixation
To evaluate the specificity and binding of the probes, in situ hybridization was performed using pure cultures of various Micromonospora and Bradyrhizobium type strains as targets. In addition, Streptomyces coelicolor A3(2) was used as a negative control. Cells were grown in ISP 2 broth (YMA for Bradyrhizobium) for 3–4 days at 28 °C, harvested and washed several times in phosphate-buffered saline 1X (PBS, 130 mM Na2HPO4, 30 mM NaH2PO4, pH 7.2). Three volumes of fresh fixation buffer (4% paraformaldehyde in PBS, pH 7.2) were added to one volume of sample (concentration adjusted to 109 cells per ml) and incubated for 3 h at 4 °C. Following fixation, cells were centrifuged and washed in PBS, harvested by centrifugation, resuspended in 1:1 (v v−1) PBS/ethanol solution. Five microliters of each fixed sample was applied to 10-well poly-L-lysine covered slides (Erie Scientific Company) and dried at 37 °C for 20 min.
Nodules were fixed in sterile 4% paraformaldehyde and 0.5% glutaraldehyde in PBS and vacuum was applied for 20 min (Speed vac, Savant) and incubated at 4 °C overnight. After washing several times with PBS 1X, the nodules were embedded in paraffin (Sakura, Tissue Tek) and sections of 2 μm were cut and mounted on 10-well poly-L-lysine coated slides and air dried. Before permeabilization, nodule sections were incubated at 60 °C for 15 min and then deparaffinized in a xylene bath (2 times, 10 min) followed by successive passages through 100, 90, 70 and 50% ethanol washes (5 min each) ending with a final wash in distilled water.
Permeabilization
To facilitate probe penetration into the cells, pure cultures and nodule sections were treated with lysozyme (5 mg ml−1) followed by proteinase K (5 μg ml−1) in 0.1M Tris-HCl, 5 mM EDTA (pH 8) for 10 min at 25 °C for each enzyme. All samples were washed in Millipore MilliQ water and dehydrated by successive passages through 50, 70, and 100% ethanol for 3 min each, air dried and processed for hybridization.
Fluorescent in situ Hybridization
For hybridization, 9 μl of hybridization solution (NaCl 0.9M, 20 mM Tris-HCl, 0.1% SDS, pH 7.4) were added to each well that contained sample (pure culture or nodule tissue), followed by 1 μl of the test probe (50 ng). Slides were placed in a chamber humidified with distilled water and incubated for 2–4 h at 48 °C. After hybridization, samples were placed in washing solution (0.05M NaCl, 0.01M Tris-HCl, 0.01% SDS, 0.05M EDTA, pH 7.4) for 20 min at 48 °C and rinsed 3 times with distilled water and air-dried.
The slides were mounted with polyvinyl alcohol mounting medium with DABCO, antifanding solution (Fluka BioChemica, Steinheim, Germany) and then observed with a confocal laser scanning microscope equipped with a neon laser Axiovert 100M). Excitation at 633 nm with emission collected between 675 and 690 nm were used to localize Cy5.5. Despite the greatly reduced sample autofluorescence with red excitation, control samples carried through the hybridization procedures without probes still had a few fluorescing spots. To eliminate this noise, these fluorescence counts were subtracted from samples with probes. Confocal settings were chosen to optimize contrast and minimize background.
Probe EU338 labelled with FITC (488 nm excitation) was observed with an epifluorescence microscope (Leica, DMRXA) to define conditions for FISH experiments.
Amplification, cloning and sequencing of the gyrB gene of Micromonospora
Total DNA was extracted from four surface sterilized, peeled lupin root nodules (each separately) using a combination of CTAB, liquid nitrogen and grinding as described by Ramírez-Saad et al. (1996). All working reagent solutions were freshly prepared and great care was taken to avoid contamination of the samples with exogenous DNA. Amplification of the gyrB gene was performed using the GYF1 (5′-TCCGGYGGYCTGCACGGCGT-3′; position 19–38) and GYR1B (5′-CGGAAGCCCTCYTCGTGSGT-3′; position 548–567) primers designed for the specific amplification of Micromonospora strains (Garcia et al., 2010). PCR amplification of a 500 bp fragment was obtained in a final volume of 20 μl using the REDExtract-N-Amp Plant PCR kit (Sigma) as described before. PCR conditions were an initial denaturation at 95 °C for 9 min, followed by 35 cycles with denaturation at 95 °C for 1 min, annealing at 62 °C for 1 min and extension at 72 °C for 2 min, followed by a step at 72 °C for 7 min. DNA from pure cultures of Micromonospora pisi DSM 45175T (Garcia et al., 2010) and Bradyrhizobium canariense BTA-1T (Rivas et al., 2009) were used as positive and negative controls, respectively. PCR products were purified using the QIAquick kit (Qiagen), ligated into the pGEM-T Easy vector (Promega, Madison, WI, USA) and cloned in Escherichia coli JM109. Plasmid purification was done with the Wizard kit (Promega) following the manufacturer's instructions. Clones containing the expected size fragment were sequenced using an automatic sequencer ABI 373A.
Microscopy
For transmission electron microscopy, nodules were cut in half along their length from the root downwards, fixed overnight in 2.5% (vol vol–1) glutaraldehyde in 0.05 M sodium cacodylate and vacuum infiltrated to remove air bubbles. The samples were subsequently stained with osmic acid, and embedded in LR White resin according to the manufacturer's instructions (The London Resin Co., Loughborough, England). Sections were cut on a Leica UC6 microtome (Leica, Milton Keynes, UK) and examined in an FEI Tecnai 20 transmission electron microscope (FEI, Eindhoven, The Netherlands) at 200 kV. For scanning electron microscopy, nodules were cut in half and fixed as described above. Samples were then dehydrated through a graded ethanol series, critical-point dried and sputter-coated with gold and examined in a Zeiss, DSM 940 scanning electron microscope.
Re-infection tests
Surface-sterilized seeds of Lupinus albus were germinated axenically in petri dishes. Seedlings were transferred to pots with sterile vermiculite and watered with nitrogen-free Rigaud and Puppo nutrient solution (Rigaud and Puppo, 1975). Fifteen plantelets (five per treatment) were inoculated with 1 ml each of a bacterial suspension (108 cells per ml) of strains Micromonospora Lupac 08 and Micromonospora Lupac 09T. The inoculated plants were placed for 10 weeks in a plant growth chamber with mixed incandescent and fluorescent lighting (400 microeinsteins m_2 s_1; 400 to 700 nm), programmed for a 16-h photoperiod, day–night cycle, with a constant temperature in the range of 25–27 °C and 50–60% relative humidity. Plants inoculated with Bradyrhizobium sp. ISLU 65 served as positive controls and uninoculated L. albus plants watered with nitrogen-free Rigaud and Puppo solution were used as negative controls.
Results
Soil characterization
The physico–chemical characteristics of the five soil samples are shown in Table 1. With respect to the texture, all samples were characterized by a high percentage of a sand fraction, especially coarse sand, resulting in the presence of sandy loam textures, which would favour drainage, aeration and biological activity. The pH of the soils ranged from 5.23 (Saelices soil) to 7.99 (Cabrerizos soil). The organic matter varied from a low of 0.20–0.40% for the two soil samples from Cabrerizos to a 1.92% content for one of the soils from Plasencia. The nitrogen values ranged from a low of 0.025% (Cabrerizos) to 0.121% (Plasencia). The inorganic phosphate content ranged from 4.54 to 8.7 mg kg−1. Available K and Mg were present in low amounts, however, important differences were found in the amount of Ca present in Plasencia (13–17 mg kg−1) and Saelices (245 mg kg−1).
Bacterial isolation and morphological characterization
One hundred and thirty-six orange pigmented, Gram-positive actinomycete-like colonies were isolated from surface sterilized root nodules on yeast mannitol agar along with Rhizobium-like bacteria after a 3-week period of incubation at 28 °C (Supplementary Figure S1). A total of 136 strains were isolated from the sampling sites: 74 colonies were counted on the isolation plates from the nodules collected in Cabrerizos. In this case nine nodules from three plants did not yield any Micromonospora strains. The final number of isolates from the remaining 20 nodules was 61 as some of the strains were lost during the purification process. Twenty-one of the 32 nodules from the plants collected in Plasencia yielded 67 Micromonospora colonies and 61 strains were obtained in pure culture. In this case no isolates were obtained from four plants. Fourteen strains were recovered from the 10 nodules collected in Saelices; four nodules from two different plants did not contain any Micromonospora. Thus, the distribution of Micromonospora in the 64 nodules sampled varied from no isolates in plants collected from the three different sampling sites to as many as 15, 22 or up to 29 colonies from a single nodule (Cabrerizos); however, in most cases, 1 to 4 colonies were obtained. Therefore Micromonospora strains were recovered in 73% of the nodules sampled.
All 136 actinobacterial strains showed a morphology described for the genus Micromonospora, they were orange, filamentous, lacked aerial mycelium and turned dark after 3 weeks upon sporulation. Fast growing microorganisms such as Aspergillus, Penicillium, Bacillus and Streptomyces were rarely observed on the isolation plates.
BOX–PCR profiles
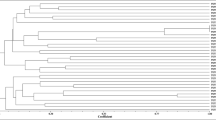
High resolution fingerprints were obtained for the 136 actinomycetes isolated from the nitrogen-fixing root nodules of L. angustifolius. The band fragments ranged from 0.1 to 2.2 kb. Clusters based on the Pearson's moment coefficient and UPGMA algorithm were defined at the 60% similarity level, yielding 37 clusters and revealing a significant genomic diversity of the isolates. Figure 1 shows the diversity of the genetic profiles for representative strains, the complete data set is available as Supplementary Material (Supplementary Figure S2). One hundred and eighteen strains were distributed in 19 clusters containing 2–15 strains; the remaining 18 isolates had a unique profile. With respect to the isolation site, the strains isolated from Plasencia (61 strains) were included in 12 clusters; the 61 strains recovered from Cabrerizos were found in 20 clusters and the 14 strains from Saelices formed 6 clusters. In one case strains from Plasencia and Cabrerizos were recovered in the same cluster (cluster 12); however, none of the 37 defined groups contained strains from the three different sampling sites. The composition of each cluster including number of strains, isolation site and code is given in Table 2.

Dendrogram showing genetic relatedness of 64 Micromonospora strains isolated from L. angustifolius determined by analysis of BOX–PCR fingerprints using the Pearson's coefficient and UPGMA cluster methods.
Multidimensional scaling analysis
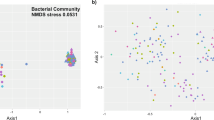
Statistical multi-dimensional scaling was performed on BOX patterns from each strain. A distribution of the isolates marked according to their geographical origin is given in Figure 2. Groups A, B, C and D contained 72% of the strains isolated from the plants collected in Cabrerizos (green dots), except for strain SB1-28, (in group D), which originated from Plasencia. A significant number of the Cabrerizos strains (21%) did not show any relationship to any of the remaining strains and appeared scattered on the plot area. The isolates from Plasencia (in red) formed three groups (E, G and I) and were well separated from the four previous groups. However, three isolates from Saelices (Lupac 02, and Lupac 04A and Lupac 04B) joined groups G and I. Six strains from Plasencia (SB1-35, SB1-32, SB1-24, SB1-13, SB1-39 and SB1-14) did not relate to any other strains and were loosely scattered. Only 5 of the 14 strains from Saelices were recovered as one moderately related group (J). Finally groups F and H contained strains isolated from the three different regions. These results point to the high diversity of the strains isolated from the nodules based on their BOX–PCR fingerprints and indicate the complexity of the Micromonospora populations isolated. In some cases, strains obtained from the same nodule did not group together (for example, MIA65 and MIA74). In other cases, strains isolated from nodules of different plants showed a close relationship (for example, groups G, H and I).

Multi-dimensional scaling analysis based on BOX–PCR fingerprints of the 136 strains isolated from root nodules collected in Cabrerizos (green), Plasencia (red) and Saelices (blue).
16S rRNA Phylogeny
Sixty-four isolates representing the 37 BOX–PCR clusters were selected for 16S rRNA gene sequencing. Nearly complete 16S rRNA gene sequences (∼1450 nt) were obtained for all strains. NCBI and Eztaxon nucleotide blast searches revealed that 100% of the sequenced microorganisms were identified with the genus Micromonospora as suggested by their morphological characteristics. Sequence similarities between the new isolates and currently described Micromonospora species ranged from 98.0 to 100%. The population evaluated showed moderate phylogenetic relationship with 11 different Micromonospora species: Micromonospora olivasteraspora (clusters 1, 2, 4, 5, 6, 16), Micromonospora coxensis (clusters 3, 11, 19, 20), Micromonospora lupini (clusters 7, 8, 9), Micromonospora coriariae (clusters 10, 27), Micromonospora aurantiaca (cluster 10), Micromonospora fulviviridis (cluster 12), Micromonospora chaiyapumensis (clusters 13, 14, 15, 37), Micromonospora echinospora (clusters 17, 21, 22, 23, 24), Micromonospora narathiwatensis (cluster 23), Micromonospora saelicesensis (clusters 28, 30, 31, 32, 33, 34, 35, 36), Micromonospora auratinigra (cluster 29) and Micromonospora sagamiensis (cluster 18). Similarity values of the sequenced strains and identification of the closest phylogenetic neighbours are given in Table 2.
The phylogenetic tree based on the neighbour-joining method (Figure 3) shows the distribution of the 64 isolates and the most closely related Micromonospora species. A tree using all presently described Micromonospora species was also constructed (not shown). In most cases, the strains grouped in the same branch as the type strains to which they were most closely related, however, a significant number of the strains did not group with any of the currently recognized species.

Neighbour-joining tree based on 16S rRNA gene sequences showing the relationships among the 64 strains sequenced and the phylogenetically closest Micromonospora species. Bootstrap percentages (from 1000 replicates) above 50% are shown at nodes. Bar, 0.005 substitutions per nucleotide position.
A significant number of the sequenced isolates (∼40%) showed <99% sequence similarity with any of the 37 Micromonospora species with validly published names. For example, isolates MIA60, MIA62 and MIA74 showed similarity values of 98.6–98.8% to M. coxensis; the similarity between the three strains was also below 99%. The branch grouping strains MIA01, MIA10, MIA03, MIA08, MIA38, MIA05 and MIA55 with sequence similarity values of 99.8–100% among them shared less than 99% similarity with M. echinospora. Isolate SB1-39, recovered in a single branch had a similarity of 98.9% with M. echinospora its closest phylogenetic neighbour. A group of 13 strains isolated from Plasencia (except MIA42), formed a complex group with sequence similarities ranging from 98.6–100%. Moderate phylogenetic relation was found with two species M. olivasteraspora and M. auratinigra (97.4–99%). This cluster grouped strains isolated from two different collection sites, Plasencia and Cabrerizos. Finally, MIA65 isolated from Cabrerizos formed a branch by itself and had a 98.6% sequence similarity with M. auratinigra. The above groups probably represent new taxa given their phylogenetic position with respect to the 37 described Micromonospora species.
Twenty-five percent of the strains isolated showed a relationship between 99–99.4% with described Micromonospora species (Table 2). These strains formed a complex branch and grouped with M. coriariae (MIA16 and MIA48), M. narathiwatensis (MIA58), and M. fulviviridis (SB1-35, SB2-2, SB2-1, SB1-4, SB1-44). However, isolates MIA50, SB2-5 and SB1-45 did not cluster with any type strain. Furthermore, taxonomic work will be necessary to determine the status of these strains.
The remaining strains (35%) had sequence similarity values higher than 99.4% with recognized species including M. chaiyaphumensis, recently described; (Jongrungruangchok et al., 2008); M. echinospora, M. coriariae, M. lupini, and M. saelicesensis; the latter three isolated from nitrogen-fixing nodules (Trujillo et al., 2006, 2007). These results strongly suggest that a significant number of different Micromonospora species are able to establish a relationship with nitrogen-fixing nodules.
Growth in nitrogen-free media
Thirty-seven strains representing all BOX–PCR clusters were chosen to determine their ability to grow in nitrogen-free media as a quick screen to select strains for nifH gene detection. Twenty-eight of the 37 strains showed good growth in semi-solid agar after a 3-week period (see Supplementary Figure S1). In some cases, the growth was similar to the growth in the control medium supplemented with ammonium sulphate.
Amplification and sequencing of the nifH gene
Based on the capacity to grow in nitrogen-free medium, representative strains were further screened for the presence of nifH genes.
Using various primer combinations PCR fragments of 337–613 bp were obtained for M. lupini Lupac 08 and M. saelicesensis Lupac 09T (Saelices); isolates SB1-39 and SB1-46 (Plasencia); and MIA32, MIA38 and MIA57 (Cabrerizos); thus, nifH-like gene sequences were obtained for representative strains isolated from the three collection sites under study. The corresponding nifH gene fragment was also amplified for the positive control, F. alni ACN14a, but not for S. coelicolor A3(2), the negative control. The sequenced fragments corresponded to the nifH gene of Frankia alni (Table 3); specifically, the strains showed a sequence similarity of 98–99% with Frankia alni ACN14a and the translated nucleotides indicated that the sequences corresponded to the dinitrogenase reductase protein (nitrogenase iron protein component II). The phylogenetic tree constructed with the neighbour-joining algorithm (Figure 4) shows the relationship of the sequences for the seven strains in this study, the sequences of the non-Frankia strains isolated from Casuarina equisetifolia (Valdés et al., 2005), and the corresponding sequences of Frankia alni ACN14a and ArI3, Frankia sp. CcI3 and Frankia sp. EAN1pec. Three groups were distinguished: group 1 formed a tight clade containing the Micromonospora strains isolated from the nodules of L. angustifolius and the two F. alni representatives ACN14a and ArI3; this branch was supported by a bootstrap value of 88% based on 1000 replicates. Group 2 supported by a bootstrap of 99% included isolates L5, L3, 7501 and Frankia sp. CcI3, isolated from Casuarina. Group 3 contained the nifH sequence of strain EAN1pec isolated from Elaeagnus.

Neighbour-joining analysis of nifH gene sequences showing the relationship among the seven isolates obtained in this study and related strains retrieved from the public databases. The significance of each branch is indicated by a bootstrap value for 1000 subsets. Bar, 0.05 substitutions per nucleotide position.
Fluorescence in situ Hybridization
Whole cell hybridizations performed with pure cultures of M. lupini Lupac 08 and Bradyrhizobium sp. ISLU 65 were carried out to show the specificities of the probes designed. The probes MicromCy5.5 and IsluCy5.5 hybridized only with Micromonospora and Bradyrhizobium, respectively. Additional FISH experiments carried out to test probe IsluCy5.5 against Micromonospora and MicromCy5.5 against Bradyrhizobium were negative (Figure 5). These results confirmed in silico simulation using the Probe Match Program (http://rdp.cme.msu.edu/probematch/search.jsp).

Pure cultures of Bradyrhizobium sp. ISLU 65 and M. lupini Lupac 08 hybridized with probes Islu Cy5.5 and Microm Cy5.5. (a) Bradyrhizobium sp. ISLU 65 hybridized with probe IsluCy5.5. Left, phase contrast; middle, hybridization signal; right, superimposed images. (b) Bradyhrizobium sp. ISLU 65 hybridized with probe MicromCy5.5. Left, phase contrast image; middle, hybridization signal; right, superimposed images. (c) M. lupini Lupac 08 hybridized with Microm Cy5.5. Left, phase contrast image; middle, hybridization signal; right, superimposed images. (d) M. lupini Lupac 08 hybridized with Islu Cy5.5. Left, phase contrast image; middle, hybridization signal; right, superimposed images. Bar, 2 μm.
Nodule sections without any probe were scanned between 420 and 620 nm to determine plant tissue autofluorescence. The fluorochrome Cy5.5 was selected over other molecules because reliable detection of bacteria on the plant tissue was only achieved when samples were excited at 633 nm, where nodule tissue autofluorescence was significantly less compared with lower excitation wavelengths. (Supplementary Figure S3)
Histological nodule sections of 2 μm were used to hibridize with probe MicromCy5.5 to detect Micromonospora inside the plant cells. Figure 6 shows bacteria-infected plant cells where intercellular spaces are also clearly seen. No fluorescence signal was dectected in infected areas where presumably Bradyrhizobium is present showing specific hybridization as previously determined by the pure culture experiments. Image analysis and confocal settings allowed elimination of autofluorescence counts originating from the plant tissue to ensure that fluorescence was due to specific hybridization with the bacteria inside the nodules. Our results indeed, indicate the presence of Micromonospora in the wild nodule tissues of L. angustifolius.

Detection of Micromonospora inside nitrogen-fixing nodules of the wild legume Lupinus angustifolius. Overlay of DIC and fluorescence image. Bar, 10 μm.
Amplification, cloning and sequencing of the gyrB gene of Micromonospora
To support the idea that Micromonospora strains were isolated from the interior of the root nodules, direct amplification of a 500 bp corresponding to an internal fragment of the gyrB gene (accession no. FN645943) was obtained from the pooled DNA isolated from the nodules. Blast comparison of the sequenced cloned amplicons indicated that two of these fragments corresponded to a gyrB gene sequence with a similarity of 98% to the environmental isolate Micromonospora sp. 37 (de Menezes et al., 2008). The next 50 sequences in the Blast list corresponded to additional gyrB gene sequences of other Micromonospora strains including validly described Micromonospora species. Therefore, correct identification was assumed. In the case of the DNA controls from Micromonospora pisi DSM 45175T and Bradyrhizobium canariense BTA-1T positive and negative results were obtained, respectively.
Microscopy
To search for the presence of Micromonospora in nodule tissues, scanning and transmission electron microscopy was done. The presence of filamentous bacteria in different parts of the nodule structure can be seen in Figure 7. The filamentous microorganism shown in Figure 7b was located underneath the first cell layers of a host nodule (Figure 7a) suggesting entrance of the bacterium. Hyphal size (0.5–0.9 μm) corresponds to bacterial not fungal hyphae. Figure 7c shows an infected nodule cell containing bacteroids and a possible hyphal structure.

s.e.m. of longitudinal section of a wild lupine nodule (a, b, c). Localization of hyphal growth on the internal tissue layers (a); amplification of a zone from the previous panel where a filamentous bacterium is near an open host cell (b), possible hyphal structure (c). TEM of filamentous-like bacterial structures entering or inside the host cells (d and f). A septum, indicated by arrows can be seen in the filamentous structure in (e). The bars in panels a, b and c are 100, 10 and 2 μm, respectively whereas those in panels d, e and f are 500 nm. x, xylem; m, mitochondria; cw, host cell wall; b, bacteroids; h?, possible hyphae; *structures which may correspond to Micromonospora.
Different plant cell organelles can be seen in all TEM photographs, in addition, the structures indicated by an asterisk (Figures 7d and f) suggest filamentous bacterial structure; they are definitely not bacteroids. These hyphal structures were located in the nodule cortex region. The density of the cytoplasm in these structures is different than that of the host cell suggesting they are not cell wall or membrane invaginations. Figure 7e shows what appears to be a septum in the filamentous structure.
Re-infection tests
Ten plants were inoculated with bacterial suspensions of Micromonospora strains Lupac 08 and Lupac 09T. After 10 weeks incubation none of the 10 plants inoculated with the two Micromonospora strains developed nodules; uninoculated control plants did not form nodules either while the plants inoculated with the Bradyrhizobium sp. ISLU 65 were nodulated.
Discussion
Bradyrhizobium is a symbiotic nitrogen-fixing soil microorganism that has the ability to form root nodules on diverse genistoid legumes. Plants of the genus Lupinus, which accomodates around 250 species of herbs and shrubs distributed in the Mediterranean area and in the American continent, are nodulated by Bradyrhizobium spp. (Jarabo-Lorenzo et al., 2003; Vinuesa et al., 2005; Stepkowski et al., 2007).
In this study, the number of Micromonospora strains recovered from the root nodules of the legumes sampled in our laboratory strongly suggests that large populations of Micromonospora inhabit the nodule tissues of healthy leguminous plants. Isolation of Micromonospora strains was successful in 73% of the nodules sampled supporting the observation that Micromonospora is a common inhabitant of nodule tissues. The distribution pattern was very complex ranging from no isolates for some nodules to as many as 29 colonies arising from a single nodule. Variation could be observed from plant to plant or even from different nodules of the same plant. Further work will be required to understand the distribution of Micromonospora in nodules and what factors determine its presence in the host.
The fact that no other microorganisms were isolated on the plates except for the nitrogen-fixing bacteria and the Micromonospora strains, indicate that the sterilization protocol was efficient as shown by the control nodules (Supplementary Figure S1).
The differences found in the physico–chemical characteristics of the rhizosphere soils where the plants developed, did not appear to determine the presence of Micromonospora in the nodules. These results suggest that the recovery of Micromonospora was limited only by the ability of the plants to develop nodules.
The presence of Micromonospora colonies already after 7–10 days incubation might indicate that colonies did not originate from surface spores, which survived the sterilization procedure; longer incubation times are usually necessary for the isolation of Micromonospora from other substrates such as soil (Kawamoto, 1989; Thawai et al., 2005). The absence of fast-growing fungi on the isolation plates was also an indication of the effectiveness of the isolation protocol considering that antifungal compounds were not added to the isolation agar. Furthermore, it would be expected that other spore-forming, faster growing microorganisms such as Bacillus or Streptomyces would appear earlier than Micromonospora, which was not the case.
BOX–PCR fingerprinting has been shown to be a useful tool to discriminate highly related strains and has been applied to study the genetic diversity of different bacterial taxa including Micromonospora (Lanoot et al., 2004; Maldonado et al., 2008; Torres et al., 2008). To get more insight into the genetic diversity of the Micromonospora strains isolated, they were analysed by BOX–PCR fingerprinting. A high degree of genetic variation was observed indicating that the strains isolated from the nodules were not clones but represented different bacterial genotypes. The 37 groups obtained provided a useful background to determine the taxonomic relationship of the strains isolated as these groups served to select strains for 16S rRNA gene sequencing.
Multi-dimensional scale analysis was used to obtain information about the diversity of the strains isolated in relation to their geographical origin and also get a better picture of the relationship among the 136 isolates based on their BOX–PCR profiles. These analyses showed that the samples collected in Cabrerizos revealed an unexpected degree of diversity as indicated by the fact that 13 strains were recovered as single member groups. These results underpin the unexpected genomic diversity of Micromonospora populations inhabiting the nodules of L. angustifolius. Although the strains isolated from Plasencia showed a lower diversity, the strains from each sampling site formed unique populations. Only a moderate relationship was found among the three sampling sites with a few exceptions. Future taxonomic studies should help to get a better picture about the number of species isolated from each site and those found in common.
16S rRNA gene sequencing and phylogenetic analyses carried out for 64 selected strains and presently described Micromonospora species pointed out that some of these strains could not be related to any species and probably represent new taxa. There is indeed strong evidence suggesting that Micromonospora type strains, genomically defined by lower than about 70% DNA–DNA reassociation values, are delineated by threshold values of almost 99.3% 16S rRNA gene sequence similarities (Kasai et al., 2000; Thawai et al., 2007; Trujillo et al., 2006, 2007; Jongrungruangchok et al., 2008; Maldonado et al., 2008). In this study, many of the strains sequenced were found in this range. Furthermore, an important number of strains had 16S rRNA gene sequence similarities below 99%. Many of these strains very likely represent new species. Indeed, further taxonomic characterization of some of these strains has resulted in the description of two new species: M. lupini and M. saelicesensis (Trujillo et al., 2007). Therefore, good congruence was found between the 16S rRNA gene data and the BOX–PCR fingerprints to select hitherto undescribed taxa in the genus Micromonospora. At this time, M. saelicesensis seems to be the most abundant species present in the nodules; however, statistical analyses and diversity indices should be applied to confirm this observation.
In the last 4 years our research group has isolated many Micromonospora strains from nitrogen-fixing nodules of different legume species. Indeed, we have found Micromonospora strains in the root nodules of every legume species sampled so far. The significant numbers of Micromonospora populations in the root nodules suggest that the presence of this microorganism is not casual, but indicates that Micromonospora has an important ecological role, which is not restricted to a single plant species. Thus, the question is: what exactly is Micromonospora doing in the legume root nodules? The presence of nifH-like gene sequences obtained from some of these strains showed a close relationship with nifH gene sequences from Frankia ACN14a. It is possible that Micromonospora also fixes nitrogen in symbiosis with its host. Whether these strains contain the complete set of genes responsible for nitrogen fixation in legumes also remains to be studied. As suggested by Valdés et al. (2005), the similarity of the nifH gene sequences of Micromonospora and Frankia suggest a lateral gene transfer between these microorganisms, which deserves further investigation as it may shed light on our understanding of their evolutionary history. In the case of the 7 Micromonospora strains in this study, the gene seems to have been transferred from a different Frankia lineage than that of strain L5. Micromonospora L5 shows a closer relationship to the nifH gene of Frankia IPNCe16 isolated from Casuarina, the plant from where strain L5 was recovered.
As Micromonospora is a sporulating microorganism, and considering the possibility that surface spores had survived the sterilization protocol, we designed specific 16S rRNA probes to determine the presence of this organism inside the nodules by fluorescent in situ hybridization. These studies showed the specificity of the probe used to detect Micromonospora as shown by the experiments carried out with pure cultures. Furthermore, an efficient protocol was developed to apply this technique successfully to plant tissues that are usually problematic due to the presence of internal structures that show fluorescence across a wide range of wavelengths. In this study, this problem was overcome by using the fluorescent molecule Cy5.5 with emission wavelengths that coincided with low levels of autofluorescence in the plant tissue.
Simultaneous detection of Micromonospora and Bradyrhizobium in the same sample was not possible because it is essential to use fluorochromes with fluorescent spectra that can be clearly differentiated to create unique signals, in our case, the wavelength working window was very narrow due to the plant autofluorescence. Nevertheless, when both probes were applied to the same sample, the fluorescence signal was stronger, filling ‘empty spaces’ that did not show a fluorescence signal when either of the two probes were used alone, suggesting the presence of Bradyrhizobium and Micromonospora together inside the infected cells.
The presence of Micromonospora within the nodule tissues of lupin is strongly supported by TEM studies, suggesting a close interaction between the host plant and the bacterium. Long filamentous and branched structures were also observed in infected cells; however, it was not possible by visual inspection to determine whether these structures were hyphae or pleomorphic bacteriods which, in many cases, enlarge up to seven times from their original bacterial rod-shape size (Oke and Long, 1999; de Maria et al., 2005). Future studies are needed to determine how Micromonospora relates to Bradyrhizobium in a tripartite interaction; a synergistic effect of both bacteria on the plant is possible (Tokala et al., 2002).
The application of specific primers to amplify a fragment that corresponded to a gyrB gene sequence of Micromonospora provided additional evidence that this bacterium inhabits the internal tissues of legume nitrogen-fixing nodules. Selection of the gyrB gene over the 16S rRNA gene was to avoid amplification of other bacterial DNA present in the nodules, that is, Bradyrhizobium or chloroplast DNA.
Re-infection experiments in our laboratory indicate that Micromonospora is not able to induce nodules on its own and that the presence of Bradyrhizobium is probably necessary for Micromonospora to penetrate the tissues; future studies will be designed to determine how Micromonospora penetrates the plant tissues, extracellular enzymes such as cellulases and xylanases produced by many of our isolates may have an important role.
The ability to produce bioactive secondary metabolites is well documented in actinobacteria and Micromonospora is no exception. Some of these novel strains were screened for new bioactive secondary metabolites, and the results obtained confirmed the production of previously undescribed molecules, designated lupinacidins A and B (Igarashi et al., 2007). These strains may have the potential to produce other bioactive molecules, which may benefit its host plant.
In summary, our studies strongly suggest that the genus Micromonospora maintains a close interaction with leguminous plants as shown by the high numbers of strains isolated. Furthermore, the presence of this microorganism does not appear to be restricted to a single legume plant species, as our results indicate that it is widely distributed (Trujillo et al., 2006, 2007; Cerda, 2008; Rodríguez, 2008; Carro, 2009; Garcia et al., 2010). This study, therefore, shows that large Micromonospora populations have been largely overlooked in this niche, the potential ecological role played by this actinobacterium in both acthinorizal and legume root nodules is yet to be unraveled.
Accession codes
References
Amann R, Ludwig W, Schleifer K . (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59: 143–169.
Bray RH, Kurtz IT . (1945). Determination of total organic and available forms of phosphorous in soils. Soil Sci 59: 39–45.
Bremner JM, Mulvaney CS . (1982). Total Nitrogen. In: Page AL, Millar RH (eds). Methods of Soil Analysis Part 2, Chemical and Microbiological Properties. American Society of Agronomy: Madison. pp 621–622.
Carro L . (2009). Avances en la sistemática del género Micromonospora: estudio de cepas aisladas de la rizosfera y nódulos de Psium sativum. Ph.D. Thesis. Universidad de Salamanca, Spain.
Cerda E . (2008). Aislamiento de Micromonospora de nódulos de leguminosas tropicales y análisis de su interés como promotor del crecimiento vegetal. Ph.D. Thesis. Universidad de Salamanca, Spain.
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK et al. (2007). EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57: 2259–2261.
Coombs JT, Franco CM . (2003). Isolation and identification of actinobacteria from surface-sterilized wheat roots. Appl Environ Microbiol 69: 5603–5608.
Coombs JT, Michelsen PP, Franco CM . (2004). Evaluation of endophytic actinobacteria as antagonists of Gaeumannomyces graminis var. tritici in wheat. Biol Control 29: 359–366.
de Maria N, de Felipe MR, Fernández-Pascual M . (2005). Alterations induced by glyphosate on lupin photosynthetic apparatus and nodule ultrastructure and some oxygen diffusion related proteins. Plant Physiol Biochem 43: 985–993.
de Menezes AB, Lockhart RJ, Cox MJ, Allison HE, McCarthy AJ . (2008). Cellulose degradation by micromonosporas recovered from freshwater lakes and classification of these actinomycetes by DNA gyrase B gene sequencing. Appl Environ Microbiol 74: 7080–7084.
Doetsch RN . (1981). Determinative methods of light microscopy. In: Gerdhardt P, Murray RGE, Costilow RN, Nester EW, Wood WA, Krieg NR, Phillips GB (eds). Manual of Methods for General Bacteriology. American Society for Microbiology: Washington. pp 21–33.
Garcia LC, Martínez-Molina E, Trujillo ME . (2010). Micromonospora pisi sp. nov., isolated from root nodules of Pisum sativum. Int J Syst Evol Microbiol 60: 331–337.
Gtari M, Brusetti L, Hassen A, Mora D, Daffoncihio D, Boudabous A . (2007). Genetic diversity among Elaeagnus compatible Frankia strains and sympatric-related nitrogen-fixing actinobacteria revealed by nifH sequence analysis. Soil Biol Biochem 39: 372–377.
Igarashi Y, Trujillo ME, Martinez-Molina E, Yanase S, Miyanaga S, Obata T et al. (2007). Antitumor anthraquinones from an endophytic actinomycete Micromonospora lupini sp. nov. Bioorg Med Chem Lett 17: 3702–3705.
Ismet A, Vikineswarg S, Paramaswari S, Wang W, Ward A, Seki T et al. (2004). Production and chemical characterization of antifungal metabolites from Micromonospora sp. M39 isolated from mangrove rhizosphere soil. World J Microbiol Biotechnol 20: 523–528.
Jarabo-Lorenzo A, Perez-Galdona R, Donate-Correa J, Rivas R, Velazquez E, Hernandez M et al. (2003). Genetic diversity of bradyrhizobial populations from diverse geographic origins that nodulate Lupinus spp. and Ornithopus spp. Syst Appl Microbiol 26: 611–623.
Jongrungruangchok S, Tanasupawat S, Kudo T . (2008). Micromonospora chaiyaphumensis sp. nov., isolated from Thai soils. Int J Syst Evol Microbiol 58: 924–928.
Kasai H, Tamura T, Harayama S . (2000). Intrageneric relationships among Micromonospora species deduced from gyrB-based phylogeny and DNA relatedness. Int J Syst Evol Microbiol 50 (Part 1): 127–134.
Kawamoto I . (1989). Genus Micromonospora Ørskov 1923, 147AL. In: Williams ST, Sharpe ME, Holt JG (eds). Bergey's Manual of Systematic Bacteriology. Williams & Wilkins: Baltimore. pp 2442–2450.
Kimura M . (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120.
Lane DJ . (1991). 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds). Nucleic acid Techniques in Bacterial Systematics. Wiley: New York. pp 115–175.
Lanoot B, Vancanneyt M, Dawyndt P, Cnockaert M, Zhang J, Huang Y et al. (2004). BOX-pCR fingerprinting as a powerful tool to reveal synonymous names in the genus Streptomyces. Emended descriptions are proposed for the species Streptomyces cinereorectus, S. fradiae, S. tricolor, S. colombiensis, S. filamentosus, S. vinaceus and S. phaeopurpureus. Syst Appl Microbiol 27: 84–92.
Maldonado LA, Stach JE, Pathom-aree W, Ward AC, Bull AT, Goodfellow M . (2005). Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie Van Leeuwenhoek 87: 11–18.
Maldonado LA, Stach JE, Ward AC, Bull AT, Goodfellow M . (2008). Characterisation of micromonosporae from aquatic environments using molecular taxonomic methods. Antonie Van Leeuwenhoek 94: 289–298.
Normand P, Simonet P, Bardin R . (1988). Conservation of nif sequences in Frankia. Mol Gen Genet 213: 238–246.
Oke V, Long SR . (1999). Bacteroid formation in the Rhizobium–legume symbiosis. Curr Opin Microbiol 2: 641–646.
Poly F, Monrozier LJ, Bally R . (2001). Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res Microbiol 152: 95–103.
Ramírez-Saad H, Akkermans WL, Akkermans DL . (1996). DNA extraction from actinorhizal nodules. In: Akkermans, ADL, van Elsas JD, de Bruijn, FJ (eds). Molecular Microbial Ecology Manual. Kluver Academic Publishers: Dordrecht. pp 1–11.
Rigaud J, Puppo A . (1975). Indole-3-acetic catabolism by soybean bacteroids. J Gen Microbiol 8: 223–228.
Rivas R, Martens M, Ladujie P, Willems A . (2009). Multilocus sequence analysis of the genus Bradyrhizobium. System Appl Microbiol 32: 101–110.
Rivas R, Sanchez M, Trujillo ME, Zurdo-Pineiro JL, Mateos PF, Martinez-Molina E et al. (2003). Xylanimonas cellulosilytica gen. nov., sp. nov., a xylanolytic bacterium isolated from a decayed tree (Ulmus nigra). Int J Syst Evol Microbiol 53: 99–103.
Rodríguez R . (2008). Análisis de la población bacteriana endofita presente en nódulos de Lupinus: interacción y localización in situ. Ph.D. Thesis. Universidad de Salamanca, Spain.
Rosenblueth M, Martinez-Romero E . (2006). Bacterial endophytes and their interactions with hosts. Mol Plant Microbe Interact 19: 827–837.
Saitou N, Nei M . (1987). The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425.
Shirling EB, Gottlieb D . (1966). Methods for characterization of Streptomyces species. Int J Syst Bacteriol 16: 313–340.
Stepkowski T, Hughes CE, Law IJ, Markiewicz L, Gurda D, Chlebicka A et al. (2007). Diversification of lupine Bradyrhizobium strains: evidence from nodulation gene trees. Appl Environ Microbiol 73: 3254–3264.
Tamura K, Dudley J, Nei M, Kumar S . (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
Thawai C, Kittakoop P, Tanasupawat S, Suwanborirux K, Sriklung K, Thebtaranonth Y . (2004). Micromonosporin A, a novel 24-membered polyene lactam macrolide from Micromonospora sp. isolated from peat swamp forest. Chem Biodivers 1: 640–645.
Thawai C, Tanasupawat S, Itoh T, Suwanborirux K, Kudo T . (2005). Micromonospora siamensis sp. nov., isolated from Thai peat swamp forest. J Gen Appl Microbiol 51: 229–234.
Thawai C, Tanasupawat S, Suwanborirux K, Itoh T, Kudo T . (2007). Micromonospora narathiwatensis sp. nov., from Thai peat swamp forest soils. J Gen Appl Microbiol 53: 287–293.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG . (1997). The clustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acid Res 24: 4876–4882.
Tokala RK, Strap JL, Jung CM, Crawford DL, Salove MH, Deobald LA et al. (2002). Novel plant-microbe rhizosphere interaction involving Streptomyces lydicus WYEC108 and the pea plant (Pisum sativum). Appl Environ Microbiol 68: 2161–2171.
Torres AR, Araujo WL, Cursino L, Hungria M, Plotegher F, Mostasso FL et al. (2008). Diversity of endophytic enterobacteria associated with different host plants. J Microbiol 46: 373–379.
Trujillo ME, Kroppenstedt RM, Fernandez-Molinero C, Schumann P, Martinez-Molina E . (2007). Micromonospora lupini sp. nov. and Micromonospora saelicesensis sp. nov., isolated from root nodules of Lupinus angustifolius. Int J Syst Evol Microbiol 57: 2799–2804.
Trujillo ME, Kroppenstedt RM, Schumann P, Carro L, Martinez-Molina E . (2006). Micromonospora coriariae sp. nov., isolated from root nodules of Coriaria myrtifolia. Int J Syst Evol Microbiol 56: 2381–2385.
Valdés M, Perez NO, Estrada-de Los Santos P, Caballero-Mellado J, Pena-Cabriales JJ, Normand P et al. (2005). Non-Frankia actinomycetes isolated from surface-sterilized roots of Casuarina equisetifolia fix nitrogen. Appl Environ Microbiol 71: 460–466.
Versalovic J, Scheider M, De Bruijn FJ, Lupski JR . (1994). Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Meth Mol Cell Biol 5: 25–40.
Vincent JM . (1970). The cultivation, isolation and maintenance of rhizobia. In: Vincent JM (eds). A Manual for the Practical Study of Root Nodule Bacteria. Blackwell Scientific: Oxford. pp 1–13.
Vinuesa P, Leon-Barrios M, Silva C, Willems A, Jarabo-Lorenzo A, Perez-Galdona R et al. (2005). Bradyrhizobium canariense sp. nov., an acid-tolerant endosymbiont that nodulates endemic genistoid legumes (Papilionoideae: Genisteae) from the Canary Islands, along with Bradyrhizobium japonicum bv. genistearum, Bradyrhizobium genospecies alpha and Bradyrhizobium genospecies beta. Int J Syst Evol Microbiol 55: 569–575.
Walkley A . (1947). A critical examination of rapid method for determining organic carbon soil-effect of variations in digestión condition and inorganic soil constituents. Soil Sci 63: 251–264.
Zakhia F, Jeder H, Willems A, Gillis M, Dreyfus B, de Lajudie P . (2006). Diverse bacteria associated with root nodules of spontaneous legumes in Tunisia and first report for nifH-like gene within the genera Microbacterium and Starkeya. Microb Ecol 51: 375–393.
Zhao H, Kassama Y, Young M, Kell DB, Goodacre R . (2004). Differentiation of Micromonospora isolates from a coastal sediment in Wales on the basis of Fourier transform infrared spectroscopy, 16S rRNA sequence analysis, and the amplified fragment length polymorphism technique. Appl Environ Microbiol 70: 6619–6627.
Acknowledgements
This work was funded by the Ministerio de Educación y Ciencia under the project CGL2006-06988 to MET and EMM. Ph.D. grants are also acknowledged to the following supporting bodies: Ministerio de Educación y Ciencia (PAV, AP20052608), Universidad de Pamplona, Colombia (RR), Junta de Castilla y León (LC), and Banco Santander (EC). We thank Carmelo Avila for his advice in statistical analyses; K. Findlay and M. Ortiz for carrying out microscopy work and Ann M. Hirsch for her valuable comments. We thank three anonymous reviewers for their constructive comments and recommendations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Trujillo, M., Alonso-Vega, P., Rodríguez, R. et al. The genus Micromonospora is widespread in legume root nodules: the example of Lupinus angustifolius. ISME J 4, 1265–1281 (2010). https://doi.org/10.1038/ismej.2010.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.55

{kind=link}
{kind=link}