
Abstract
As a major cyanophage group, cyanobacterial podoviruses are important in regulating the biomass and population structure of picocyanobacteria in the ocean. However, little is known about their biogeography in the open ocean. This study represents the first survey of the biodiversity of cyanopodoviruses in the global oceans based on the viral encoded DNA polymerase (pol) gene. A total of 303 DNA pol sequences were amplified by PCR from 10 virus communities collected in the Atlantic and Pacific oceans and the South China Sea. At least five subclusters of cyanopodoviruses were identified in these samples, and one subcluster (subcluster VIII) was found in all sampling sites and comprised approximately 50% of total sequences. The diversity index based on the DNA pol gene sequences recovered through PCR suggests that cyanopodoviruses are less diverse in these oceanic samples than in a previously studied estuarine environment. Although diverse podoviruses were present in the global ocean, each sample was dominated by one major group of cyanopodoviruses. No clear biogeographic patterns were observed using statistical analysis. A metagenomic analysis based on the Global Ocean Sampling database indicates that other types of cyanopodovirus-like DNA pol sequences were present in the global ocean. Together, our study results suggest that cyanopodoviruses are widely distributed in the ocean but their community composition varies with local environments.
Similar content being viewed by others


Introduction
Viruses are believed to be the most abundant component of the marine microbial communities (Bergh et al., 1989; Suttle, 2005) and are critical in enhancing geochemical cycling in the microbial loop and mediating horizontal gene transfer (Fuhrman, 1999; Wilhelm and Suttle, 1999; Wommack and Colwell, 2000; Weinbauer, 2004). Nearly 20 years ago, Proctor and Fuhrman (1990) reported that 1–3% of the cyanobacteria from diverse marine locations contained mature viral particles. Since then, viruses have been isolated using different groups of marine and freshwater picocyanobacteria, including Synechococcus and Prochlorococcus strains (Suttle and Chan, 1993; Waterbury and Valois, 1993; Wilson et al., 1993; Suttle, 2000; Lu et al., 2001; Sullivan et al., 2003; Sullivan et al., 2005; Wilhelm et al., 2006; Pope et al., 2007; Wang and Chen, 2008). It has been estimated that cyanophages may be responsible for approximately 5–14% of cyanobacterial mortality on a daily basis (Suttle and Chan, 1994; Binder 1999). A recent metagenomic study on oceanic bacterial community showed that cyanophage-related sequences represented up to 10% of total fosmid sequences and 60–80% of virus sequences (DeLong et al., 2006). Another metagenomic analysis targeting the viral fraction also showed that cyanophages were dominated in the Sargasso Sea and widespread in marine environments (Angly et al., 2006). These observations imply a dynamic interaction between cyanophage and cyanobacteria in the ocean.
All isolated cyanophages have been classified into three-tailed phage families: the Myoviridae, Podoviridae and Siphoviridae (Suttle, 2000). Typically, cyanomyoviruses are able to cross-infect a broad range of hosts, whereas cyanopodoviruses and cyanosiphoviruses are more host specific (Sullivan et al., 2003; Weigele et al., 2007; Wang and Chen, 2008). The genetic diversity of cyanomyoviruses in various aquatic environments has been explored extensively based on their conserved capsid gene (g20) (Fuller et al., 1998; Zhong et al., 2002; Wang and Chen, 2004; Filee et al., 2005; Short and Suttle, 2005; Wilhelm et al., 2006; Sullivan et al., 2008). In contrast, little is known about the diversity of cyanopodoviruses and cyanosiphoviruses.
It is believed that cyanopodoviruses are important due to their high host specificity and rapid production. A recent study showed that podoviruses infecting marine picocyanobacteria contain a conserved DNA polymerase gene (DNA pol), which can be used as a gene marker to investigate the genetic diversity of these viruses (Wang and Chen, 2008). PCR amplification, cloning and sequencing of this viral DNA pol gene from field samples have uncovered new genotypes and subclusters of cyanopodoviruses in the Chesapeake Bay (Chen et al., 2009). However, given that the population structure of picocyanobacteria (the host organisms) in the coastal estuary is different from that in the open ocean (Chen et al., 2006), it has become important to survey the diversity of this group as little is known about the biogeography of the cyanopodoviruses in the open ocean. In this study, we used the same PCR primer set to explore the genetic diversity of viral communities across the global oceans, including five samples from the North Atlantic Ocean, four from the Pacific Ocean and one from the South China Sea.
Materials and methods
Water sample collection
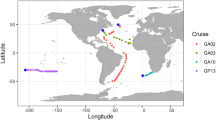
Nine water samples (Figure 1) were collected from the surface waters of the North Atlantic Ocean in May and June 2005 (on the R/V Seward Johnson), and from the Pacific Ocean in January 2007 (on the R/V Kilo Moana) and September 2008 (on the R/V Tangaroa). All water samples were collected using Niskin bottles attached to the ships’ rosette systems. Twenty liters of water was prefiltered through 142-mm diameter GF/F filters (Whatman, Clifton, NJ, USA) and then through 0.45- and/or 0.22-μm nominal pore-size 142-mm diameter polycarbonate filters (Millipore, Bedford, MA, USA). The virus-size fraction was then concentrated to ∼500 ml using Millipore's M12 ProFlux tangential flow filtration with a 30 kDa cutoff filter cartridge (S10Y30; Millipore). One water sample, SCS1, was collected from the surface water of the South China Sea in November 2006 (on the R/V Dong Fang Hong II) (Figure 1). Water was prefiltered through 198-mm diameter 3-μm pore-size filters (Millipore) and viral concentrates were condensed from 50 l to a final volume of 1 l on board by the same kind of tangential flow filtration facility. All the virus concentrates were stored in the dark at 4 °C. Temperature, salinity and the concentration of chlorophyll a at all the sampling sites are shown in Table 1.

The locations of sampling sites. Ten locations were indicated by pink solid circles with their station names beside. Sampling sites were localized by using Surfer software (Golden Graphics, Golden, CO, USA). The base-map was an annual composite of chlorophyll a concentration in 2005 obtained from the NASA website (http://oceancolor.gsfc.nasa.gov/).
DNA extraction and PCR amplification
Owing to the relatively low viral abundance in open ocean samples, we modified the protocol in our previous study (Chen et al., 2009). First, we used extracted viral community DNA as template instead of boiled viral concentrate. Second, a touchdown PCR program with a lower annealing temperature was used. Viral particles in the VCs (1 ml) were treated with 0.5 mg ml−1 (final concentration) proteinase K for 3 h, followed by a phenol–chloroform protocol. DNA was precipitated by isopropyl alcohol and resolved in 30 ml TE buffer. A set of PCR primers designed in previous research of the DNA polymerase gene of cyanopodoviruses in Chesapeake Bay (Chen et al., 2009) was used in this study. The forward primer is CP-DNAP-349F (5′-CCAAAYCTYGCMCARGT-3′), and the reverse primer CP-DNAP-533R consists of equal molar amounts of two primers 533Ra (5′-CTCGTCRTGSACRAASGC-3′) and 533Rb (5′-CTCGTCRTGDATRAASGC-3′). All PCR reactions were performed in 50 μl volume containing 1 × reaction buffer, 2.5 mM MgCl2, 100 mM dNTPs, 10 pmol of each primer, 1 U of Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA, USA) and 2 μl DNA solutions for templates. A touchdown PCR program was performed, which consisted of an initial step of denaturing at 94 °C for 3 min, followed by 32 cycles of denaturing at 94 °C for 30 s, annealing, for 30 s, extension at 72 °C for 1 min and a final extension at 72 °C for 10 min. The annealing temperature was decreased from 49 by 0.5 °C every cycle for first four cycles, followed by 28 cycles at 47 °C. The reaction was performed in the PTC-200 Peltier Thermal Cycler (MJ Research, Waltham, MA, USA). With these modifications, about 78% of oceanic samples (21 of 27) yielded a positive amplification (10 samples are presenting in this study; data not shown for others).
Cloning, sequencing and phylogenetic analysis
PCR products were purified using UltraClean PCR Clean-up DNA Purification Kit (Mo Bio Laboratories, Solana Beach, CA, USA) and cloned using the TOPO TA Cloning Kit (Invitrogen) both by following the manufacturer's instructions. About 30 clones of each library were sequenced using BigDye terminator chemistry and an ABI 3100 Genetic Analyzer (PE Applied Biosystems, Foster City, CA, USA) at the Center of Marine Biotechnology, UMBI. The translated amino-acid sequences were aligned using ClustalX2 (Larkin et al., 2007), and the alignment was modified using MEGA 4.0 software (http://www.megasoftware.net/) (Tamura et al., 2007) manually. Neighbor-joining (NJ) trees of amino-acid sequences were constructed by using MEGA 4.0 with the default parameters. Maximum likelihood (ML) analysis was conducted by using PHYML program (http://www.lirmm.fr/guindon/phyml.html) (Guindon and Gascuel, 2003) under the WAG substitution matrix. Tree topologies generated by both methods were broadly similar. Bootstrap resamplings were performed for 1000 and 100 replications in NJ and ML analyses. The sequences in this study were deposited in GenBank (accession numbers GU082016–GU082318).
Multidimensional scaling and diversity analyses
Nonmetric multidimensional scaling (MDS) analysis was performed using SPSS (version 13.0) (SPSS Inc, Chicago, IL, USA) software to determine the visually perceptual distances between clone libraries based on the phylogenetic clustering results shown in Table 2. Distance between DNA sequences of libraries was pairwise calculated by using the DNADIST program of PHYLIP (http://evolution.genetics.washington.edu/phylip.html). The distances were served as input files to determine the diversity indices. Operational taxonomic units and abundance-based coverage estimator of each library were measured using software DOTUR (http://schloss.micro.umass.edu/software/dotur.html) based on DNA sequences at 2% divergence.
Metagenomic analysis
Selected deduced amino-acid sequences of cyanopodovirus DNA pol gene from all the subclusters were BLAST searched against two microbial community metagenome databases: the Global Ocean Sampling (GOS; Rusch et al., 2007; Williamson et al., 2008) and the HOT/ALOHA (DeLong et al., 2006). They are two components of the CAMERA database (http://camera.calit2.net/; Seshadri et al., 2007). Both GOS and HOT metagenomic studies showed that cyanophage-related sequences are commonly present in their databases. The BLAST search was also conducted against the marine virioplankton metagenome database (Angly et al., 2006), but the results were not used here because the hits were not reliable and the sequences were too short (∼100 bp) for the phylogenetic analysis. The optimized BLAST E-value was set at ⩽10−20. There were 479 BLAST hits from the GOS database but no hits from HOT. The GOS viral DNA pol-like sequences were aligned with our sequences. Only those sequences that were fully overlapped with our PCR sequences and fell within the cyanopodovirus cluster were included in the phylogenetic tree.
Results and Discussion
Phylogenetic diversity of viral DNA pol sequences in the ocean
A total of 303 DNA pol gene sequences were recovered from the 10 ocean samples, and they were distributed into five subclusters (II, III, VIII, IX and XI) with 90% amino-acid sequence identify cutoff for each subcluster (Figure 2a). The majority (93%) of ocean DNA pol sequences (283 of 303) were confined to three major subclusters (II, VIII and XI). A large portion (approximately 50%) of oceanic DNA pol sequences (153 clones) fell into subcluster VIII (Figure 3). Sequences in subcluster VIII were present in all open ocean samples, and dominated samples UTK211, UTK220, UTK240, UTK295 and SCS1 (Table 2). A closer examination of subcluster VIII shows a great deal of sequence heterogeneity, which can be divided into at least three main subpopulations (VIII-A, VIII-B and VIII-C; Figure 2b). Members in VIII-A and VIII-B were common in the open oceans, whereas the VIII-C members were dominant in Chesapeake Bay. It is noteworthy that the majority of Chesapeake Bay sequences in subcluster VIII were found during the winter when the bay was largely influenced by the offshore water (Chen et al., 2009). Subcluster VIII is an interesting phylogenetic group because it includes a large pool of widely distributed cyanopodoviurses that have limited host identity. Recently, the DNA pol gene sequences from three podoviruses infecting marine Synechococcus WH8101 were determined, and these fell into subcluster VIII (M Marston, personal communication), suggesting that isolation and characterization of cyanophages from diverse cyanobacterial hosts are critical toward a comprehensive understanding of cyanophage diversity in the ocean.


Phylogenetic relationships of DNA pol gene recovered from environmental samples and cyanopodovirus isolations (a). All the environmental sequences in the previous study about the Chesapeake Bay (Chen et al., 2009) were appended into the alignments as references. The sequences from eight cyanopodovirus isolates and the partial sequences amplified from the sediment samples in Strait of Georgia and water column samples of Gulf of Mexico (Labonté et al., 2009) are shown in black. The sequences recovered from open ocean, Chesapeake Bay and GOS samples are in blue, green and pink, respectively. The numbers of clones or sequences within a subcluster are given in parentheses. A closed-up view of DNA pol sequence diversity within subcluster VIII (b). Bootstrap values (>50%) for maximum likelihood (ML) and distance analyses are shown on the main branches in the order of ML/NJ. The inconsistent branches between ML and distances analyses are indicated by thinner dotted lines. The scale bars represent 0.1 and 0.01 amino-acid substitutions per site for the lager and small trees, respectively.

The comparison of distribution frequency of DNA pol subclusters between Chesapeake estuary (221 sequences) and open ocean samples (303 sequences).
Eighty-one sequences (26.7%, 81 of 303) from the ocean samples were closely related to podovirus P60, and formed the second largest lineage, subcluster XI (Figure 2a). Sequences in subcluster XI were recovered from six oceanic samples, among which three samples (UTK229, UTK250 and UTK255) were dominated by sequences in this subcluster (Table 2). P60 is a lytic phage that infects Synchococcus WH7805 (Chen and Lu, 2002), a cyanobacterium isolated from the Atlantic Ocean (Waterbury et al., 1986). P60 was recognized as a highly active phage because of its short latent period compared to other cyanophages (Wang and Chen, 2008). All the oceanic sequences in this subcluster shared greater than 97% amino-acid identity with P60, suggesting that P60-like podoviruses are common in the ocean and could be critical in regulating the population dynamics of oceanic cyanobacteria. The majority of sequences recovered from samples UTK202 and UKT262 were in subcluster II (Table 2). Till date, no cyanopodovirus representative falls into this subcluster.
Clone library analysis shows lower genetic diversity of cyanopodoviruses in the open oceans compared to an estuary
The composition of DNA pol genotypes in the open oceans is different from that in the Chesapeake Bay (Figure 3). For example, nearly 40% of DNA pol sequences in the Chesapeake estuary were in subcluster I, but no subcluster I members were found in the open oceans. In contrast, members of subcluster IX were found in the open ocean, but not in the Chesapeake Bay. The DNA pol sequences from ocean samples were grouped into five subclusters, whereas the Chesapeake Bay sequences were divided into 10 subclusters. Almost all the subclusters (except for subcluster IX) for the ocean samples can be found in the Chesapeake Bay. In an early study conducted in the Chesapeake Bay, a similar PCR protocol was used to amplify the cyanopodovirus DNA pol gene from eight samples, and approximately 30 clones were sequenced for each sample (Chen et al., 2009). On the basis of comparison of clone libraries between open oceans and the Chesapeake Bay, cyanopodoviruses in the open ocean appear to be less diverse compared with those in the estuary. The low diversity of cyanopodoviruses in the open ocean was also supported by the rarefaction curves (Figure 4) and richness index (Table 2). The average abundance-based coverage estimator richness index is 15.4 and 52.3 for the ocean samples and Chesapeake Bay samples. All nine UTK ocean samples were dominated by a specific group of cyanopodoviruses (>60% clone libraries) and had richness index values less than 14.0 (Table 2). The South China Sea sample has a relatively high richness index value (64.8).

Rarefaction curves of 10 offshore clone libraries. Two selected Chesapeake Bay clone libraries (Chen et al., 2009) that represent the winter and summer viral communities are shown as references.
High genetic diversity of cyanopodoviruses in the coastal estuary could be related to the complexity of their hosts. Synechococcus in coastal or mesotrophic oceanic waters are more abundant (Partensky et al., 1999) and diverse (Zwirglmaier et al., 2008) than those in oligotrophic oceans. Because cyanopodoviruses are highly host specific, the low abundance and diversity of Synechococcus populations in open ocean waters may partially account for the low diversity (and possibly low abundance) of Synechococcus cyanopodoviruses. The primer set used in this study was able to amplify the DNA pol gene from podoviruses either P-SSP7 (Sullivan et al., 2005) or Syn5 (Pope et al., 2007), which infect marine Prochlorococcus MED4 and Synechococcus WH8109, respectively (data not shown). However, no clonal sequence closely related to P-SSP7 and Syn5 was obtained from our ocean samples. Podoviruses infecting high-light-adapted Prochlorococcus have been isolated from ocean's surface water (Sullivan et al., 2003). It is not clear whether the absence of P-SSP7-like Prochlorococcus podoviruses in this study is due to the low viral abundance or the bias of PCR primers. Currently, only one DNA pol sequence from Prochlorococcus podovirus is available in GenBank. Clearly, more DNA pol sequences from Prochlorococcus podoviruses are warranted to better understand the global diversity of cyanopodoviruses.
The MDS analysis showed no distinct geographic patterns for oceanic cyanopodoviruses (Figure 5). Nine UTK libraries were clustered into three groups, whereas the South China Sea sample SCS1 was distantly discriminated on the MDS map. Samples from different locations of Pacific and Atlantic oceans (that is, UTK229, UTK250 and UTK255) could share high similarity of clone library composition despite the great variability in the concentration of chlorophyll a and water temperature. For instance, the clustering samples UTK211, UTK220, UTK240 and UTK295 were from sites with a broad water temperature range (9–20 °C) and chlorophyll range (0.08–1.8 μg per liter) (Table 1), but they were grouped together (Figure 5). In this case, no clear geographic distribution pattern was seen based on the chlorophyll level or temperature gradient. The similarity observed for the viral DNA pol sequences between the Atlantic and Pacific oceans suggests that other unknown factors may have a key role on the distribution of cyanopodoviruses. The ubiquitous distribution of picocyanobacteria in the open oceans may partially explain the similarity of viral composition between the two major oceans. Because podoviruses infect specific hosts, it is plausible that cyanopodoviruses may correspond to cyanobacterial distribution geographically, and such a hypothesis remains to be tested in the future.

MDS (multidimensional scaling) plots showing the distribution pattern of 10 cyanopodovirus community libraries. The MDS analysis was performed based on the phylogenetic clustering shown in Table 2. Sampling stations were represented by empty circles. Stations UTK211, UTK220 and UTK295 were clustered at the same location on the MDS map indicated by a bold circle. Samples with relatively close distance on the map are grouped by dashed circles.
Metagenomes of host-derived cyanopodoviruses revealed unknown viral groups
When selected amino-acid sequences from subclusters I–XI were blasted against the GOS database, 479 sequences were retrieved with the E-value cutoff at 10−20. A subset (191) of 479 GOS sequences overlapped with our sequences recovered from environmental samples in full length. Among these 191 sequences, 84 clustered within the cyanopodovirus regime whereas 107 clustered with the T7 group (data not shown). All the 84 sequences analyzed here came from 31 GOS sites. The GOS sequences were clustered into seven groups (Figure 2a). ‘Groups’ were used for the GOS sequences because they were not supported by high bootstrap values (that is, GOS unknown group II, V and VI). Among the seven GOS groups, one group that contains eight GOS sequences was closely related to Prochlorococcus podovirus P-SSP7. The remaining six GOS groups represented cyanopodoviruses with unknown host origin. Surprisingly, little overlap was seen between our ocean sequences and the GOS sequences (except for GOS unknown group I). The discrepancy between these two data sets could be caused by different sampling locations, sample preparation or PCR bias. Regardless, the metagenomic analysis suggests that other unknown groups of cyanopodoviruses are present in the open ocean, increasing the global diversity of cyanopodoviruses dramatically.
One PCR amplicon sequence (SCS1-131) from the South China Sea sample fell into GOS group I. Meanwhile, there were three other SCS sequences (SCS1-123, 105 and 106) clustered with GOS shotgun sequences. Unlike the UTK ocean samples, the SCS sample was prefiltered through a 3 μm pore-size filter instead of 0.2 or 0.45 μm ones. Therefore, the SCS sample contains microbial assemblages including viruses, bacteria and other small plankton (<3 μm). The PCR amplicon from the SCS sample likely includes the cyanopodovirus DNA pol sequences from both free and host-derived viral communities. The other five GOS unknown groups (II–VI) were unique compared with the PCR-derived sequences. It is noteworthy that two groups of GOS DNA pol sequences compose 43 sequences, forming unknown group III (22 sequences) and IV (21 sequences). Both groups are widely distributed across 12 GOS sites. Members of GOS unknown group IV appear to be affiliated with three DNA pol sequences (SOG-S-D, SOG-S-L and SOG-S-G) found in sediments of Strait of Georgia (Labonté et al., 2009). According to the DNA pol phylogeny (Labonté et al., 2009), these three SOG sequences formed a unique cluster and had no homologue with GOS sequences. However, we were able to retrieve 18 GOS sequences that are closely related to the viral pol sequences from the Strait of Georgia sequences, suggesting that this group of viruses is widely distributed in different environments.
It was unexpected that there were only three GOS sequences in subcluster VIII. Members of subcluster VIII dominated our oceanic samples. It is noted that all the GOS samples mainly include microbes with size ranging from 0.1 to 0.8 μm. Picocyanobacteria such as marine Synechococcus could be poorly represented by this size fraction because of their relatively large cell size. This size fraction concern was previously reported in other studies in that less than 10% cyanobacteria-like scaffolds generated from Sargasso Sea shotgun sequencing seemed to be derived from Synechococcus (Venter et al., 2004). GOS study also showed that Synechococcus recruited 4–6 times less reads than Prochlorococcus across global oceans (Rusch et al., 2007). Whether the low appearance frequency of GOS sequences in subcluster VIII is caused by sampling bias is not clear.
We blasted the representative DNA pol sequences against both viral and bacterial community genomes (that is, viromic, GOS and HOT metagenomic database). The viromic database (Angly et al., 2006) contains short sequences (approximately 100 nt or 33aa), which could not provide reliable hits or long sequences for the phylogenetic analysis. The GOS and HOT metagenome databases contain long sequence reads (approximately 1000 bp), which are critical for BLAST search and phylogenetic analysis. The downside of blasting viral genes against the GOS and HOT metagenomic databases is that they were not set up originally for viral communities. Viral sequences only make up approximately 3% of the total predicted proteins of GOS database (Williamson et al., 2008). Cyanophage sequences comprise up to 10% of HOT microbial metagenome (DeLong et al., 2006), but no cyanopodovirus DNA pol gene sequences were found in the data set. Different cloning and sequencing strategies and magnitudes of sequences between GOS and HOT samples may result in this discrepancy.
Conclusion
This study shows that podoviruses of picocyanobacteria are widely distributed in the world's oceans. The comparison of clone libraries suggests that cyanopodoviruses in the open oceans are less diverse (and perhaps less abundant) compared to those in the estuarine ecosystem. A large portion of cyanopodoviruses (subcluster VIII) in the ocean appears to be ubiquitous and represents a potentially important ecological group. Metagenomic analysis of GOS host-derived DNA pol sequences suggests that cyanopodoviruses are diverse across global oceans. Although podoviruses are typically virulent and host specific, the interaction between podoviruses and cyanobacteria in the ocean still remains largely unknown. Co-monitoring of biogeography of both picocyanobacteria and their viruses is warranted to further understand the ecological role of cyanophage in the marine environment.
Accession codes
References
Angly F, Felts B, Breitbart M, Salamon P, Edwards R, Carlson C et al. (2006). The marine viromes of four oceanic regions. PLoS Biol 4: e368.
Bergh O, Borsheim KY, Bratbak G, Heldal M . (1989). High abundance of viruses found in aquatic environments. Nature 340: 467–468.
Binder B . (1999). Reconsidering the relationship between virally induced bacterial mortality and frequency of infected cells. Aquat Microb Ecol 18: 207–215.
Chen F, Lu JR . (2002). Genomic sequence and evolution of marine cyanophage P60: a new insight on lytic and lysogenic phages. Appl Environ Microbiol 68: 2589–2594.
Chen F, Wang K, Kan JJ, Suzuki MT, Wommack KE . (2006). Diverse and unique picocyanobacteria in Chesapeake Bay, revealed by 16S-23S rRNA internal transcribed spacer sequences. Appl Environ Microbiol 72: 2239–2243.
Chen F, Wang K, Huang SJ, Cai HY, Zhao MR, Jiao NZ et al. (2009). Diverse and dynamic populations of cyanobacterial podoviruses in the Chesapeake Bay unveiled through DNA polymerase gene sequences. Environ Microblol 11: 2884–2892.
DeLong EF, Preston CM, Mincer T, Rich V, Hallam SJ, Frigaard NV (2006). Community genomics among stratified microbial assemblages in the ocean's interior. Science 311: 496–503.
Filee J, Tetart F, Suttle CA, Krisch HM . (2005). Marine T4-type bacteriophages, a ubiquitous component of the dark matter of the biosphere. Proc Natl Acad Sci USA 102: 12471–12476.
Fuhrman JA . (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399: 541–548.
Fuller NJ, Wilson WH, Joint IR, Mann NH . (1998). Occurrence of a sequence in marine cyanophages similar to that of T4 g20 and its application to PCR-based detection and quantification techniques. Appl Environ Microbiol 64: 2051–2060.
Guindon S, Gascuel O . (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systemat Biol 52: 696–704.
Labonté J, Reid KE, Suttle CA . (2009). Phylogenetic analysis indicates evolutionary diversity and environmental segregation of marine podovirus DNA polymerase gene sequences. Appl Environ Microbiol 75: 3634–3640.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H et al. (2007). ClustalW2 and ClustalX version 2. Bioinformatics 23: 2947–2948
Lu J, Chen F, Hodson RE . (2001). Distribution, isolation, host specificity, and diversity of cyanophages infecting marine Synechococcus spp. in the Georgia river estuaries. Appl Environ Microbiol 67: 3285–3290.
Partensky F, Blanchot J, Vaulot D . (1999). Differential distribution and ecology of Prochlorococcus and Synechococcus in oceanic waters: a review. In: Charpy L and Larkum AWD (eds). Marine Cyanobacteria. Bulletin de l’Institut oceanographique de Monaco, No. spécial 19, Musée océanographique: Monaco, MONACO, pp 457–475.
Pope WH, Weigele PR, Chang J, Pedulla ML, Ford ME, Houtz JM et al. (2007). Genome sequence, structural proteins, and capsid organization of the cyanophage Syn5: a ‘horned’ bacteriophage of marine Synechococcus. J Mol Biol 368: 966–981.
Proctor LM, Fuhrman JA . (1990). Viral mortality of marine bacteria and cyanobacteria. Nature 343: 60–62.
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S et al. (2007). The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol 5: e77.
Seshadri RS, Kravitz A, Smarr L, Gilna P, Frazier M . (2007). CAMERA: a community resource for metagenomics. PLoS Biol 5: e75.
Short CM, Suttle CA . (2005). Nearly identical bacteriophage structural gene sequences are widely distributed in both marine and freshwater environments. Appl Environ Microbiol 71: 480–486.
Sullivan MB, Waterbury JB, Chisholm SW . (2003). Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature 424: 1047–1051.
Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW . (2005). Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol 3: e144.
Sullivan MB, Coleman ML, Quinlivan V, Rosenkrantz JE, DeFrancesco AS, Tan G et al. (2008). Portal protein diversity and phage ecology. Environ Microbiol 10: 2810–2823.
Suttle CA, Chan AM . (1993). Marine cyanophages infecting oceanic and coastal strains of Synechococcus: abundance, morphology, cross-reactivity and growth characteristics. Mar Ecol Prog Ser 92: 99–109.
Suttle CA, Chan AM . (1994). Dynamics and distribution of cyanophages and their effect on marine Synechococcus spp. Appl Environ Microbiol 60: 3167–3174.
Suttle CA, Chen F . (1992). Mechanisms and rates of decay of marine viruses in seawater. Appl Environ Microbiol 58: 3721–3729.
Suttle CA . (2000). Cyanophages and their role in the ecology of cyanobacteria. In: Whitton BA and Potts M (eds). The Ecology of Cyanobacteria: Their Diversity in Time and Space. Kluwer: Boston, pp 563–589.
Suttle CA . (2005). Viruses in the sea. Nature 437: 365–361.
Tamura K, Dudley J, Nei M, Kumar S . (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304: 66–74.
Wang K, Chen F . (2004). Genetic diversity and population dynamics of cyanophage communities in the Chesapeake Bay. Aquat Microb Ecol 34: 105–116.
Wang K, Chen F . (2008). Prevalence of highly host-specific cyanophages in the estuarine environment. Environ Microbiol 10: 300–312.
Waterbury JB, Valois FW . (1993). Resistance to co-occurring phages enables marine Synechococcus communities to coexist with cyanophages abundant in seawater. Appl Environ Microbiol 59: 3393–3399.
Waterbury JB, Watson SW, Valois FW, Franks DG . (1986). Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus. Can Bull Fish Aquat Sci 214: 71–120.
Weigele PR, Pope WH, Pedulla ML, Houtz JM, Smith AL, Conway JF et al. (2007). Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ Microbiol 9: 1675–1695.
Weinbauer MG . (2004). Ecology of prokaryotic viruses. FEMS Microbiol Rev 28: 127–181.
Wilson WH, Joint IR, Carr NG, Mann NH . (1993). Isolation and molecular characterization of five marine cyanophages propagated on Synechococcus sp. strain WH7803. Appl Environ Microbiol 59: 3736–3742
Wilhelm SW, Carberry MJ, Eldridge ML, Poorvin L, Saxton MA, Doblin MA . (2006). Marine and freshwater cyanophages in a Laurentian Great Lake: evidence from infectivity assays and molecular analyses of g20 genes. Appl Environ Microbiol 72: 4957–4963.
Wilhelm SW, Suttle CA . (1999). Viruses and nutrient cycles in the sea. Bioscience 49: 781–788.
Williamson SJ, Rusch DB, Yooseph S, Halpern AL, Heidelberg KB, Glass HI et al. (2008). The Sorcerer II global ocean sampling expedition: metagenomic characterization of viruses within aquatic microbial samples. PLoS One 1: e1456
Wommack KE, Colwell RR . (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev 64: 69–114.
Zhong Y, Chen F, Wilhelm SW, Poorvin L, Hodson RE . (2002). Phylogenetic diversity of marine cyanophage isolates and natural virus communities as revealed by sequences of viral capsid assembly protein gene g20. Appl Environ Microbiol 68: 1576–1584.
Zwirglmaier K, Jardillier L, Ostrowski M, Mazard S, Garczarek L, Vaulot D et al. (2008). Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol 10: 147–161.
Acknowledgements
We thank Janet Rowe and Audrey Matteson for assistance in sample collection; M Marston for providing the information about newly isolated cyanopodoviruses; M Sullivan, J Waterbury and F Valois for providing cyanophage lysates of P-SSP7 and Syn5, respectively. We also thank Wan-Hsin Chen for editing the article. This project was in part supported by the Xiamen University 111 program (to FC) and 2007CB815904 (to NZJ), and NSF grants OCE-0452409 and OCE-0825405 (to SWW).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Huang, S., Wilhelm, S., Jiao, N. et al. Ubiquitous cyanobacterial podoviruses in the global oceans unveiled through viral DNA polymerase gene sequences. ISME J 4, 1243–1251 (2010). https://doi.org/10.1038/ismej.2010.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.56
Keywords
This article is cited by
-
Abundant and cosmopolitan lineage of cyanopodoviruses lacking a DNA polymerase gene
The ISME Journal (2023)
-
Molecular Diversity of Cyanopodoviruses in Two Coastal Wetlands in Northeast China
Current Microbiology (2019)
-
Cyanophage-encoded lipid desaturases: oceanic distribution, diversity and function
The ISME Journal (2018)
-
Genetic diversity and temporal dynamics of phytoplankton viruses in East Lake, China
Virologica Sinica (2015)
-
Shotgun metagenomics indicates novel family A DNA polymerases predominate within marine virioplankton
The ISME Journal (2014)
