
Abstract
Genes of the major histocompatibility complex (MHC) encode receptor molecules that are responsible for recognition of intracellular and extracellular pathogens (class I and class II genes, respectively) in vertebrates. Given the different roles of class I and II MHC genes, one might expect the strength of selection to differ between these two classes. Different selective pressures may also promote different rates of gene conversion at each class. Despite these predictions, surprisingly few studies have looked at differences between class I and II genes in terms of both selection and gene conversion. Here, we investigated the molecular evolution of MHC class I and II genes in five closely related species of prairie grouse (Centrocercus and Tympanuchus) that possess one class I and two class II loci. We found striking differences in the strength of balancing selection acting on MHC class I versus class II genes. More than half of the putative antigen-binding sites (ABS) of class II were under positive or episodic diversifying selection, compared with only 10% at class I. We also found that gene conversion had a stronger role in shaping the evolution of MHC class II than class I. Overall, the combination of strong positive (balancing) selection and frequent gene conversion has maintained higher diversity of MHC class II than class I in prairie grouse. This is one of the first studies clearly demonstrating that macroevolutionary mechanisms can act differently on genes involved in the immune response against intracellular and extracellular pathogens.
Similar content being viewed by others

Introduction
The major histocompatibility complex (MHC) has an essential role in the immune response of vertebrates. MHC genes encode receptor molecules that bind small fragments of peptides derived from the processing of pathogens and present them to immune cells, initiating a cascade of immune reactions (Janeway et al., 2005). There are two major subgroups of MHC genes, class I and class II, which are primarily responsible for the recognition of intracellular and extracellular pathogens, respectively (Janeway et al., 2005). MHC genes are considered the most polymorphic loci in the vertebrate nuclear genome (Hughes, 1999). In humans, over 8000 MHC allelic variants have been resolved worldwide with nearly 1800 allelic variants at the MHC DRB-1 locus alone (Robinson et al., 2013), and nucleotide diversity in the MHC region is up to two orders of magnitude higher than the genomic average (Garrigan and Hedrick, 2003). In birds, hundreds of alleles in each class have been reported for some species, for example, 862 class I and 977 class II alleles were identified in the great tit Parus major (Sepil et al., 2012) and common yellowthroat Geothlypis trichas (Bollmer et al., 2012), respectively. MHC polymorphism at the species or population level is thought to be primarily driven by the immense diversity of parasites and pathogens (Spurgin and Richardson, 2010), and at least two major evolutionary mechanisms are responsible for maintaining this unusually high diversification of MHC alleles: positive (balancing) selection and recombination.
Balancing selection on the vertebrate MHC acts primarily through negative frequency-dependent, overdominant and fluctuating selection (Hughes and Nei, 1988; Takahata and Nei, 1990; Hedrick, 2002). Signatures of balancing selection acting on MHC genes are usually supported by three lines of evidence: (i) high allelic diversity, (ii) higher rates of non-synonymous (dN) to synonymous (dS) nucleotide substitutions than expected under neutral evolution and (iii) alleles retained between species following divergence for longer evolutionary periods than expected for non-selected loci (Kamath and Getz, 2011). If ancestral alleles are retained through speciation events, then some alleles will be more similar between species than within species (that is, trans-species polymorphism; Klein et al., 1993, 1998; Cutrera and Lacey, 2007).
MHC haplotypes may arise via meiotic reciprocal recombination (crossing over) or gene conversion, and the latter process is thought to explain most MHC allelic diversity (Hosomichi et al., 2008). There are two types of gene conversion commonly occurring in the MHC region: (i) allelic (intra-locus) gene conversion occurring during meiosis as a consequence of the repair of mismatched heteroduplexes (Galtier et al., 2001) and (ii) non-allelic (inter-locus) gene conversion between duplicated (paralogous) DNA fragments (Ohta, 1999). While de novo MHC sequence variation is generated by point mutation, gene conversion can generate new haplotypes by transferring sections of DNA within and across duplicated MHC loci (Spurgin et al., 2011). Gene conversion (either intra- or inter-locus) is now recognized to have a crucial role in generating and maintaining MHC diversity in many vertebrates (Reusch and Langefors, 2005; Schaschl et al., 2006), including birds (Chaves et al., 2010; Spurgin et al., 2011; Promerová et al., 2013).
MHC evolution is also driven by gene duplication, which produces a varying number of MHC loci in different taxa (Kelley et al., 2005), with up to 20 expressed class II B genes per individual reported for some avian species (Bollmer et al., 2010). Despite the frequent duplication events at the MHC, the birth-and-death model of MHC evolution assumes that many duplicated genes may become dysfunctional or deleted (Nei et al., 1997; Nei and Rooney, 2005). Gene loss (death) seems a possible evolutionary scenario for the surprisingly compact structure of the MHC region in Galliformes. The ‘minimal essential’ MHC was first described in the domestic chicken Gallus gallus, which has two class I and two class II B genes encoding only one dominantly expressed locus at each class (Kaufmann et al., 1999). In chickens, the MHC is dense with short introns and there are few duplications or pseudogenes, which produces a simple genetic architecture (Kaufmann, 1999). Other galliform species, such as the ring-necked pheasant Phasianus colchicus (Wittzell et al., 1999a) and black grouse Tetrao tetrix (Strand et al., 2007; Wang et al., 2012), share the compact genetic organisation of the chicken MHC. Recent studies of the greater prairie chicken Tympanuchus cupido have revealed an even simpler MHC architecture, with only one class I locus and two class II loci, leading to the smallest core MHC-B of any bird species yet studied (Eimes et al., 2013).
Although the evolution of the MHC has been the subject of research for over five decades (Clarke and Kirby, 1966; Ejsmond et al., 2014), most studies have focused on the MHC of a single species and typically a single gene, either class I or II (reviewed in Bernatchez and Landry, 2003). Thus, comparisons of MHC evolution at both class I and II are restricted to relatively few vertebrates, mostly teleost fish (Shum et al., 2001; Kruiswijk et al., 2005; Schaschl and Wegner, 2007) where class I and class II genes are distributed on different chromosomes and not contained within a single complex. Given the different roles of class I and II MHC genes and the pathogens they recognize, one might expect selection to differ between these two classes. Furthermore, recombination is expected to be different depending on the number of loci. For example, species such as the greater prairie chicken with a single class I locus and two class II loci can only show signs of intra-locus gene conversion within the single class I locus, while both intra- and inter-locus gene conversion can occur at class II where there are two loci (Reusch and Langefors, 2005). In addition, the rate of intra-locus gene conversion may depend on the strength of selective pressures, as stronger balancing selection might favour new alleles produced by gene conversion (Wegner, 2008). So far, there is limited evidence for contrasting evolution of both MHC classes in higher vertebrates, where all MHC genes are linked in a cluster on the same chromosome (Klitz et al., 1986; Bonneaud et al., 2004; Kuduk et al., 2012). Here, we investigated the molecular evolution of MHC class I and class II sequences in five closely related species of Centrocercus and Tympanuchus grouse (subfamily Tetraoninae). To date, the arrangement and number of genes in the 61 kb MHC class II B region of these species has only been determined in T. cupido (Eimes et al., 2013), and was found to have a simple MHC architecture relative to most galliform and other bird species. In this study, we sought to determine whether a simple MHC structure extended to other grouse (Tetraoninae). We focused our study on exon 3 of MHC class I genes and exon 2 of MHC class II B genes, both corresponding to the antigen-binding regions of MHC molecules. The main goals of this paper were to: (i) characterize levels of polymorphism of both MHC classes; (ii) characterize key evolutionary mechanisms (balancing selection, gene conversion) acting on MHC genes; and (iii) compare the role of these mechanisms in shaping the evolution of MHC class I and class II in grouse.
Materials and methods
Sample collection
We sampled five species of North American prairie grouse: Centrocercus minimus (Gunnison sage-grouse), C. urophasianus (greater sage-grouse), Tympanuchus phasianellus (sharp-tailed grouse), T. pallidicinctus (lesser prairie chicken) and T. cupido (greater prairie chicken). Tympanuchus and Centrocercus are closely related and, together with Dendragapus, form a monophyletic clade within the Tetraoninae (Stein et al., 2015). Blood and muscle tissue samples were collected between 1997 and 2011 for 183 greater prairie chickens and for 20–22 individuals from each of the other grouse species. Sampling locations are summarized in Supplementary Table S1. Protocols used for DNA extraction are described in previous publications (Bellinger et al., 2003; Oyler-McCance et al., 2005; Galla and Johnson, 2015).
Amplification and 454 pyrosequencing
We genotyped MHC class I exon 3 and class II B exon 2 in 266 and 263 birds, respectively (Table 1). Although these exons correspond to only ~50% of the antigen-binding regions of MHC class I and II molecules, they are known to be hypervariable (Wittzell et al., 1999b) and most MHC studies have focused exclusively on these regions (for example, Kruiswijk et al., 2005; Schaschl et al., 2006; Cutrera and Lacey, 2007; Alcaide et al., 2013, 2014). For MHC class I, we used primers 1a2inExon3F and 1a2Raltintron2 originally developed for the greater prairie chicken (Bateson et al., 2014). As previously documented for this species (Bateson et al., 2014), we observed sequence length polymorphism at exon 3 of class I in all three Tympanuchus species. The most common sequence length was 258 bp, while sequences of 255 and 261 bp were generated by a 3-bp deletion at position 58 and a 3-bp insertion at position 40, respectively. There was no sequence length polymorphism within Centrocercus (all sequences were 255 bp). For MHC class II, we used a forward primer (Blex2F) developed for greater prairie chicken (Eimes et al., 2010) and a reverse primer (RNA R1a) developed for black grouse (Tetrao tetrix, Strand et al., 2007), which amplified a 237-bp fragment of the 270-bp exon 2. PCR protocols are described elsewhere (see Bateson et al., 2014 for class I, see Eimes et al., 2010 for class II).
We used 454 pyrosequencing for genotyping both MHC class I and class II in all individuals, except for nine C. minimus that were part of an initial set that was cloned and sequenced to test the primers (see Bateson et al., 2014 for cloning methods). Amplifications were completed using fusion primers containing Roche (Basel, Switzerland) 454 adapter sequences, an 8-bp barcode used to distinguish individuals and a pair of either the MHC class I or II primers. PCR products were purified, pooled and sequenced on a Roche 454 FLX Genome Sequencer using Titanium chemistry at Research and Testing Laboratory, LLC (Lubbock, TX, USA).
MHC allele validation
Reads containing the complete forward primer, complete barcode and at least 10 internal bases of the reverse primer were extracted from multifasta files and sorted according to barcodes with jMHC software (Stuglik et al., 2011). Barcodes, adapters and primers were trimmed and low quality reads were removed. True alleles were identified in several steps. First, we extracted sequence variants of expected exon sizes for MHC class I (255, 258, 261 bp; Bateson et al., 2014) and class II (237 bp; Eimes et al., 2010). We also extracted amplicons that showed minor sequence variation (1–3 bp) from a high copy number variant of the expected size (putative true allele). Repeatable putative errors believed to originate during 454 sequencing were identified using the criteria proposed by Lighten et al., (2014) and were clustered with similar high copy number sequences for further analyses. Next, we retained only those sequence variants that were found in at least three reads (following Radwan et al., 2012), as it is extremely unlikely that three identical sequences occur by chance owing to sequencing errors (Galan et al., 2010).
We used the degree of change and copy number variation approaches developed by Lighten et al. (2014) to identify true alleles and discard all other types of artefacts. The degree of change approach estimates a number of allelic copies of each individual by differentiating putative true sequences and artefacts on the basis of the observed sequencing depths of the 10 most common sequence variants (Lighten et al., 2014). The method relies on the assumption that there should be an obvious reduction in sequencing depths between putative alleles and artefacts, so the least amplified true allele is associated with an inflection point in a linear plot of cumulative sequencing depths. The copy number variation approach identifies the number of allelic copies by comparing the observed proportion of reads for each allele within individual to a theoretical genetic model that calculates expected read counts for a range of loci and allelic copies (Lighten et al., 2014). All putative true alleles identified with either of these two approaches were checked to determine whether they could be explained as chimeras that are generated repeatedly during PCR by recombination among pairs of true alleles (Lenz and Becker, 2008). The algorithm used to identify PCR-derived chimeras from true recombinants included two criteria: (i) PCR-derived chimeras should always co-occur with both parental sequences in the same amplicon, whereas true recombinants may or may not co-occur with one or both parental sequences; (ii) PCR-derived chimeras should generally be less abundant than true alleles (following Zagalska-Neubauer et al., 2010). Identification of true alleles was confirmed by their alignment with previously published greater prairie chicken alleles (Eimes et al., 2010, 2011, 2013) in a neighbour-joining tree using Geneious v7.1.7 (http://www.geneious.com, Kearse et al., 2012).
After this processing, we had 155 807 class I and 264 253 class II reads for all individuals, with an average of 606 class I (range: 25–3124) and 1036 class II (range: 145–6197) reads per individual. On average (±s.e.), there were 347.8±15.1 and 439.7±15.2 reads per true allele within individuals, at class I and II, respectively. Genotyping repeatability of MHC class I and II was high (R=0.93; F35, 36=29.13; P<0.001), based on 18 birds with rare alleles that were repeated using independent PCRs in two separate 454 pyrosequencing runs.
Identifying locus-specific MHC class II alleles
As our 454 pyrosequencing primers amplified both MHC class II loci in prairie grouse (BLB1 and BLB2, Eimes et al., 2013), we modified the locus-specific approach of Strand et al. (2013) to identify alleles occurring at BLB1 (see Supplementary Methods). This nested PCR approach consisted of an initial amplification (2068 bp) from Blec1 to the beginning of exon 2 in BLB1 (Eimes et al., 2013). The product from this initial PCR was used as template for a second PCR that amplified exon 2 of BLB1 (see Supplementary Methods for details). To determine whether a particular allele occurred at BLB1, we genotyped five individuals (or as many as possible if a smaller sample size was available) that carried each of the MHC class II alleles identified in our sample of prairie grouse; this included 75 of 263 individuals. We used Geneious to align the sequences from the nested PCR to the alleles previously found in the same individuals with 454 pyrosequencing. We were unable to amplify the second MHC class II locus (BLB2) across prairie grouse using the long-range primers of Strand et al. (2013) or redesigned primers on the basis of our map of the greater prairie chicken MHC (Eimes et al., 2013). However, we observed a maximum of two alleles per individual with the nested PCR approach, thereby providing no evidence to suggest that we amplified more than the BLB1 locus.
Analysis of polymorphism and selection
MHC class I and class II sequences were aligned in Geneious v7.1.7 and sequence polymorphism (number of segregating sites, total number of mutations, average number of nucleotide differences and average nucleotide diversity) across all five grouse species was analysed with DnaSP v5.0 (Librado and Rozas, 2009). To test whether positive selection historically operated on the MHC class I and class II sequences in grouse, we used two different approaches. First, we compared relative rates of non-synonymous (dN) and synonymous (dS) substitutions across the sequences (Hughes and Nei, 1988). Substitution rates were computed in MEGA v6.0 (Tamura et al., 2013) according to the modified Nei-Gojobori (mNG) method (Nei and Gojobori, 1986; Zhang et al., 1998) with the ratio of the numbers of transitions and transversions (R)=2 and with the Jukes and Cantor (1969) correction for multiple substitutions at the same site. Calculations were carried out separately for the putative non-antigen-binging sites (non-ABS) and antigen-binding sites (ABS) at class I (Kaufman et al., 1992) and class II (Brown et al., 1993), including three additional ABS sites (codons 17, 48, 58) recognized by Bondinas et al. (2007) for MHC class II. A codon-based two-tailed Z-test of selection was performed separately on ABS and non-ABS regions, as well as on the entire sequence, to test for the null hypothesis of neutrality (dN/dS=1). We also used one-tailed Z-tests to determine whether positive selection (dN/dS>1) acted on ABS regions and on the entire sequence of MHC class I exon 3 and MHC class II exon 2.
Second, signatures of positive selection acting on ABS and non-ABS regions were detected with the partitioning approach for robust inference of selection (PARRIS; Scheffler et al., 2006), as implemented in the Datamonkey web server (Delport et al., 2010). The PARRIS method infers positive selection acting on nucleotide sequences by using likelihood ratio tests (LRT) to compare a null-model (M1, no-selection) where dN is restrained to be ⩽dS, against a full model (M2, selection) where dN is allowed to be >dS. This method expands on other maximum likelihood methods for detecting positive selection by allowing substitution rates to vary across sites and accounting for recombination.
We also tested for codon-specific signatures of positive and negative selection across MHC class I and class II sequences using three different approaches implemented in the Datamonkey server: single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL) and random effects likelihood (REL; Kosakovsky Pond and Frost, 2005). We considered a codon to be evolving under selection when it was identified as such by at least two methods (Winternitz and Wares, 2013). To visualize the magnitude of dN versus dS, we plotted normalized dN—dS values calculated with the FEL approach, which under most scenarios is intermediate between the SLAC and REL methods in terms of type I error (Kosakovsky Pond and Frost, 2005). We used mixed effects model of evolution (MEME; Murrell et al., 2012) to detect evidence of episodic diversifying selection acting on individual residues. This method combines fixed effects on the level of a site with random effects at the level of branches and has superior performance over previous models under a broad range of scenarios (Murrell et al., 2012).
Gene conversion
Although sequence analysis is unlikely to successfully distinguish between gene conversion and meiotic reciprocal recombination, the latter process is usually invoked to explain exchanges of large DNA segments between MHC regions (Cullen et al., 2002; Otting et al., 2005; Hosomichi et al., 2008). In the chicken, large genomic segments (>40 kb) shared between MHC haplotypes were argued to originate from reciprocal recombination, while sharing of smaller segments (1–2 kb) was attributed to gene conversion (Hosomichi et al., 2008). As we only analysed recombination at the intra-exonic level (within a 261-bp class I sequence and 239-bp class II sequence), it seems safe to assume that we were likely to detect mostly gene conversion events, and thus all recombination processes are hereafter referred to as gene conversion.
We assessed gene conversion at MHC class I and class II within and between Centrocercus and Tympanuchus with GENECONV v1.81a (Sawyer, 1989). The software detects gene conversion events by testing whether the nucleotide substitutions observed between a set of DNA sequences are randomly distributed along the sequences (Drouin et al., 1999). This approach is considered one of the most powerful to infer gene conversion events correctly among several different recombination detection methods (Posada, 2002), although it might have limited power when the conversion rate is high (Mansai and Innan, 2010). Simulated global P values <0.05 based on 10 000 permutations were considered evidence of gene conversion. Mismatches within conversion tracts were not allowed by setting the g-scale parameter (mismatch penalty) to 0. The minimum number of recombination (gene conversion) events was also calculated with the four-gamete method of Hudson and Kaplan (1985) implemented in DnaSP software. Sequence alignments of class I exon 3 and class II exon 2 were also screened for gene conversion by testing whether the physical distance between sites correlated with the measures of linkage disequilibrium (LD). For this purpose, we used two indices of LD, r2 of Hill and Robertson (1968) and D’ of Lewontin (1964), which were calculated in DnaSP. Our null hypothesis predicted that, in the absence of gene conversion, adjacent sites should show similar levels of LD to more distant sites (Meunier and Eyre-Walker, 2001; Kiemnec-Tyburczy et al., 2012), which was tested with the Pearson product-moment correlation coefficient.
Phylogenetic clustering
To assess the phylogenetical relationships of MHC class I and II in prairie grouse, we constructed phylogenetic trees using maximum likelihood (ML) in MEGA v6.0 (Tamura et al., 2013). Evolutionary distance was computed with the Hasegawa-Kishino-Yano (HKY) model and clade support was tested with 1000 bootstrap replications. Sequences of the mallard Anas platyrhynchos (GeneBank accession nos.: JN810924 for class I, AF390589 for class II), chicken Gallus gallus (GeneBank accession nos.: KF032370 for class I, HQ203710 for class II) and helmeted guineafowl Numida meleagris (GeneBank accession nos.: EF643463 for class I, EU826064 for class II) were used as outgroups.
Results
Allelic diversity
We found 37 class I and 46 class II alleles across all five grouse species (Table 1). There were more alleles in Tympanuchus cupido (25 class I and 30 class II alleles) than in the other grouse species (3–12 class I alleles; 2–15 class II alleles), but this was due to the much larger sample size for T. cupido (n=182–3) than the other species (n=19–22; Table 1). To produce comparable estimates of MHC diversity, we resampled MHC variation in T. cupido using the sample size comparable with that for the other species (n=20). Number of alleles at both class I and II was similar for all Tympanuchus species after resampling T. cupido (Table 1). There was a high rate of allele sharing within genera, with 52% (n=13) of class I and 60% (n=18) of class II alleles shared between T. cupido and the two other Tympanuchus species. Within Centrocercus, C. minimus shared all three class I alleles and one of two class II alleles with C. urophasianus. Only one class I (Tycu-IA*33) and one class II (Tycu-BLB*14) allele were shared between Centrocercus and Tympanuchus. In all species, we found one or two class I alleles per individual (mean 1.61±0.22 (s.d.); Table 1), while there were one to four class II alleles per individual in all Tympanuchus and 1–3 class II alleles per individual within Centrocercus (see Table 1), confirming the presence of only one class I locus and two class II loci in both grouse genera.
Of the 46 alleles found at class II, we confirmed the presence of 35 alleles at the BLB1 locus by genotyping 75 individuals with BLB1-specific primers after 454 pyrosequencing. For this BLB1-specific analysis, we tested all 46 class II alleles and found an average of 4.02±0.55 copies per allele across the 75 individuals. Of the 46 alleles, 24 were found only at BLB1, 11 alleles were shared between the two class II loci (BLB1 and BLB2; inferred by comparing sequences obtained with BLB1-specific primers and our previous 454 pyrosequencing results) and the other 11 alleles presumably belonged to the BLB2 locus because they were not observed in the nested BLB1 PCR results. Most of the alleles that were not found at BLB1 (9 out of 11 alleles) were relatively rare (present in <3% of individuals genotyped with 454 pyrosequencing, n=263). Below we present the results comparing class I and class II using all alleles for class II (that is, at both BLB1 and BLB2; n=46), as well as the alleles that occurred at the single BLB1 locus (n=35).
At class I, we found a lower proportion of segregating sites (39/261 segregating sites per total number of sites) and fewer mutations (η=52) than at class II (95/237 segregating sites and η=128 mutations at BLB1 alone; 96/237 segregating sites and η=131 mutations across all class II sequences (BLB1 and BLB2)). The average number of nucleotide differences was 12.97±2.13 (s.d.) for class I and 33.43±3.43 for BLB1 class II locus (31.36±3.30 for all class II sequences). Consistent with the lower mutation rate, the average nucleotide diversity was more than two times lower for class I (0.051±0.004) than class II (0.141±0.008 for BLB1 sequences; 0.132±0.008 for all class II sequences). ABS regions had much higher nucleotide diversity than non-ABS regions at both class I (ABS, 0.115±0.011 versus non-ABS, 0.038±0.003) and class II (ABS, 0.272±0.023 versus non-ABS, 0.085±0.009 for BLB1 sequences; ABS, 0.260±0.013 versus non-ABS, 0.080±0.008 for all class II sequences).
Tests of selection
There was strong support for positive selection (dN/dS>1) acting on ABS regions at both class I and II (Table 2). Although dN/dS was higher for the class I ABS region, this was mainly due to few synonymous mutations (dS), which greatly elevated the ratio despite a relatively low value of dN. By contrast, dN—dS indicated stronger selection acting on class II ABS region (Table 2). Separate analyses for each species confirmed that selection was stronger on the ABS region of class II than class I in three of four species (Supplementary Table S2; C. minimus was not analysed because of a low number of alleles at each class). Strong positive selection at ABS regions was confirmed using PARRIS as implemented in Datamonkey (LRT=14.08, P<0.001 for class I; LRT=16.77, P<0.001 for BLB1 sequences; LRT=18.07, P<0.001 for all class II sequences). A partitioning approach indicated no evidence for positive selection at non-ABS regions (LRT=4.58, P=0.10 for class I; LRT=0.00, P=1.00 for BLB1 sequences, LRT=0.04, P=0.97 for all class II sequences). There was also evidence of positive selection on the entire class II exon 2 (dN/dS=1.25 for BLB1 sequences, dN/dS=1.40 for all class II sequences), but not for class I exon 3 (dN/dS=0.94; Table 2).
The codon-based maximum likelihood approach detected signatures of positive and episodic diversifying selection acting on MHC class I less frequently than on MHC class II, regardless of comparing only BLB1 alleles or all class II sequences (Table 3; Figure 1). Fewer cases of positive or episodic diversifying selection were found at amino acid residues in MHC class I (3 of 87 residues, 3.4%) than class II (14 of 79 residues, 17.7%; G=4.88, df=1, P=0.027; Figure 1; Table 3). In general, signatures of positive or episodic diversifying selection were detected more frequently in ABS regions, when compared with non-ABS regions. There was evidence for these types of selection on 52.0% of all residues predicted to be in ABS regions of class II (n=25), in contrast to only one non-ABS site (1.9%, n=54; G=14.79, df=1, P<0.001; Figure 1, Table 3). This difference was much less pronounced and non-significant in MHC class I (G=1.65, df=1, P=0.20), with only two ABS codons (11.8%, n=17) and one non-ABS codon (1.4%, n=70) identified as under positive or episodic diversifying selection (Figure 1, Table 3). Signatures of negative (purifying) selection were more frequent in non-ABS regions, with zero or two ABS codons identified as under purifying selection at MHC class I and class II, respectively, compared with three non-ABS codons at each class (Table 3).

Alignments of amino acid sequences of MHC class I exon 3 (a) and MHC class II exon 2 (b) of Centrocercus and Tympanuchus grouse; subsets containing 15 representative alleles are shown for each MHC class. MHC class II alleles not found at BLB1 locus are in frames. Dots indicate the same amino acid with top sequence. Grey shading indicates putative ABS according to Kaufman et al. (1992) for MHC class I and according to Brown et al. (1993) and/or Bondinas et al. (2007) for MHC class II. Amino acid residues under positive selection are marked with a plus (+), under negative selection are marked with a dash (−) and under episodic diversifying selection are marked with an asterisk (*). Spatial variation in the selection parameter (normalized dN—dS) is shown at the top of each alignment.
Gene conversion
We found little evidence for gene conversion at MHC class I. In Tympanuchus, only 13% (4/32) of class I alleles had signatures of gene conversion (four significant pairwise gene conversions recognized in total, see Supplementary Figure S1 and Supplementary Table S3) and a minimum of ρM=5 gene conversion events were recognized within this genus with the four-gamete test. Centrocercus had no evidence for gene conversion at MHC class I.
The occurrence of gene conversion was much more frequent across all class II sequences. In Centrocercus, 67% (8/12) of class II alleles had signatures of gene conversion (six significant pairwise gene conversions; Supplementary Table S4), and a minimum of ρM=9 conversion events were estimated (n=12 alleles). There were four different conversion tracts (26–171 bp) identified within Centrocercus. In Tympanuchus, 91% (32/35) of class II alleles were involved in at least one intra-generic gene conversion event and 22 different conversion tracts were identified (37–183 bp). In total, the substitution approach recognized 102 significant pairwise conversions among Tympanuchus MHC class II alleles (Supplementary Figure S1 and Supplementary Table S3). The four-gamete test identified a minimum of ρM=11 conversion events. All alleles private to either T. phasianellus or T. pallidicinctus shared fragments with T. cupido alleles, and the only allele private to C. minimus shared a fragment with two C. urophasianus alleles suggesting pre-speciation conversion events. The only allele shared between Tympanuchus and Centrocercus (Tycu-BLB*14) showed evidence of conversion with one C. urophasianus allele (Ceur-BLB*11), but with no private Tympanuchus alleles, suggesting that the conversion event likely occurred after the two genera had diverged.
An analysis using only BLB1 alleles supported frequent gene conversion at MHC class II. In Centrocercus, 40% (4/10) of BLB1 alleles had signatures of gene conversion, while 88% (23/26) of BLB1 alleles were involved in at least one conversion event in Tympanuchus (2 and 62 significant pairwise gene conversions, respectively; Supplementary Table S5). Among BLB1 alleles, there were at least 8 and 11 gene conversion events (ρM) recognized with the four-gamete test in Centrocercus and Tympanuchus, respectively. After restricting the analysis to BLB1 alleles that were not shared with the BLB2 locus (7 Centrocercus alleles and 17 Tympanuchus alleles), no gene conversion events were found in Centrocercus, but there was evidence for frequent gene conversions in Tympanuchus (all 17 alleles involved in 53 significant pairwise gene conversions; Supplementary Table S6). A minimum number of gene conversions (ρM) among BLB1-specific alleles was estimated at 3 and 10 events in Centrocercus and Tympanuchus, respectively.
We also found considerable support for gene conversion at the inter-generic level, although it was less frequent in MHC class I than in class II. Gene conversion events between three private Centrocercus and four private Tympanuchus alleles (three different conversion tracts and six significant pairwise conversions, Supplementary Table S2) were detected for MHC class I, while there was evidence for gene conversion between eight Centrocercus and eight Tympanuchus private MHC class II alleles (seven different conversion tracts and 12 significant conversions; Supplementary Table S2).
These contrasting patterns of gene conversion at MHC class I and class II loci were supported by LD analysis. We found no correlation between LD statistics and physical distance between sites at MHC class I (r=0.03, P=0.62 for Lewontin D’; r=−0.03, P=0.54 for Hill and Robertson r2). By contrast, LD decreased with distance between sites across all class II sequences (r=−0.07, P<0.001 for Lewontin D’; r=−0.13, P<0.001 for Hill and Robertson r2), as well as across BLB1 sequences only (r=−0.06, P=0.005 for Lewontin D’; r=−0.12, P<0.001 for Hill and Robertson r2), indicating strong signatures of gene conversion.
Phylogenetic clustering
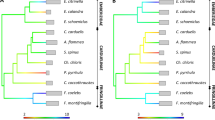
Phylogenetic trees of MHC class I and class II sequences revealed contrasting evolutionary histories (Figure 2). All MHC class I alleles of Centrocercus clustered within a single clade, yet paraphyletic with an allele shared with T. cupido (Tycu-IA*33) and one private allele of T. phasianellus (Typh-IA*02; Figure 2a). This result is consistent with the low signal of inter-generic gene conversion at MHC class I (see Figure 2a). All four private Tympanuchus alleles identified as having undergone gene conversion with Centrocercus alleles (Tycu-IA*16, Tycu-IA*23, Tycu-IA*31, Typa-IA*02) were placed in a neighbouring Tympanuchus clade (Figure 2a). By contrast, MHC class II alleles of Centrocercus grouse did not form a distinct clade and were located among other grouse species, most likely reflecting high rates of inter-generic gene conversion (Figure 2b). In fact, most Centrocercus class II alleles closely clustered with Tympanuchus alleles that were identified as having undergone inter-generic gene conversion (Figure 2b). Neither MHC class I and class II sequences formed monophyletic groups corresponding to species, which, in accordance with the high rate of allele sharing within genera, indicate trans-species evolution of this gene family in grouse.

Maximum likelihood phylogenetic trees of MHC class I exon 3 (a) and MHC class II exon 2 (b) sequences from five Centrocercus and Tympanuchus grouse. Bootstrap values are provided for nodes with >50% support, and the scale bar indicates genetic distance in units of nucleotide substitutions per site. Centrocercus alleles are shaded in grey and alleles shared between species are indicated by acronyms given in brackets (Ceur=C. urophasianus; Cemi=C. minimus; Tycu=T. cupido; Typa=T. pallidicinctus; Typh=T. phasianellus). Private Tympanuchus alleles identified as having undergone inter-generic gene conversion are marked with black squares. The mallard Anas platyrhynchos, domestic chicken Gallus gallus and helmeted guineafowl Numida meleagris were used as the outgroups (for GenBank accession numbers and references see Methods).
Discussion
We found striking differences in selection and gene conversion between MHC class I and class II genes within prairie grouse. Although we found positive selection acting on antigen-binding regions of both classes, only two putative ABS residues of class I (11.8%) were positively selected, compared with over half of putative class II ABS residues, which were under positive or episodic diversifying selection. Consequently, we found evidence of positive selection acting on the entire MHC class II exon 2, but no evidence of positive selection on the entire MHC class I exon 3. We also identified gene conversion as an important mechanism shaping the evolution of MHC class II in prairie grouse. By contrast, there was little evidence for gene conversion events occurring between class I alleles. Overall, the combination of strong positive (balancing) selection and frequent gene conversion has maintained higher diversity of MHC class II than class I in prairie grouse.
Surprisingly few studies have compared the relative importance of balancing selection on MHC class I and class II (Shum et al., 2001; Siddle et al., 2007; Kuduk et al., 2012), and only fragmentary information is available for birds. Previous studies on MHC variation in T. cupido suggested that the strength of balancing selection might be much less pronounced at class I than at class II (Eimes et al., 2013). However, this conclusion was based on the analysis of a severely bottlenecked population and it was argued that drift could have removed most evidence of selection at class I (Eimes et al., 2013). Although MHC class II variation in the same population was significantly reduced following the bottleneck, signals of historical positive selection on class II ABS codons were still retained (Eimes et al., 2011). This pattern suggests that persistence of divergent MHC class II alleles through the bottleneck was likely to be an adaptive process that preserved differences in pathogen-recognition capacity among haplotypes to support immune response, while the loss of variation at class I could be driven more by stochastic processes, such as genetic drift, rather than selection. Consistent with our results, substitution rates (dN/dS ratios) supported stronger selection for diverse MHC class II than for class I alleles in the domestic chicken (Hosomichi et al., 2008) and in a more distantly related non-passerine, the blue petrel Halobaena caerulea (Strandh et al., 2011).
A stronger signal of balancing selection at MHC class II genes is, however, unlikely to be a general pattern across avian species. Comparison of MHC class I and class II evolution within the genus Falco revealed that both classes accumulated synonymous and non-synonymous mutations at similar rates, indicating no differences in the intensity of adaptive evolution (Gangoso et al., 2012). The opposite pattern of selection can be inferred from multiple studies on two passerine species, the red-winged blackbird Agelaius phoeniceus and house finch Carpodacus mexicanus, which showed stronger signals of selection in the antigen-binding regions of MHC class I than of class II (Edwards et al., 1998; Jarvi et al., 2004; Alcaide et al., 2013). Differences in selection on the MHC class I of passerines and non-passerines may be related to their effective population sizes and exposure to pathogens (Alcaide et al., 2013), but these differences are unlikely to explain the stronger selection we found on class II than class I loci within the same set of species. Instead, our data suggest that differences in selection in prairie grouse species might be explained by stronger selection from extracellular (on class II) than intracellular (class I) pathogens.
Consistent with this hypothesis, field studies suggest that extracellular parasites are a strong selective force in wild grouse populations (reviewed in Peterson, 2004). The proventricular nematode Dispharynx nasuta causes significant mortality in the ruffed grouse Bonasa umbellus (Gross, 1925) and in the blue grouse Dendrapagus obscurus chicks (Bendell, 1955). It was also suggested to be an important factor in the extinction of the heath hen T. c. cupido (Gross, 1928). Two other nematodes, Trichostrongylus cramae and T. tenuis, cause caecal lesions and inflammation and have been associated with reduced fecundity and survival of grouse. For example, experimental reductions in the intensity of Trichostrongylus parasites led to increased body weight, adult survival, egg hatchability and nesting success in the red grouse Lagopus lagopus scotica (Shaw, 1990; Hudson et al., 1992). In prairie chickens, it has also been argued that cestode (Raillietina variabilis) infections can affect host population dynamics (Leigh, 1941; Harper et al., 1967). Similarly, nearly 50% of sampled greater sage-grouse have been reported to be infected by Raillietana tapeworms, which were suggested to have a direct or indirect adverse impact on individual birds (Christiansen and Tate, 2011). Although such infectious agents as bronchitis and reticuloendotheliosis viruses are also known to be pathogenic for prairie grouse (Drew et al., 1998; Peterson et al., 2002), in general, there is much less empirical evidence for the detrimental effects of intracellular pathogens on wild grouse populations (Peterson, 2004).
There was also a much stronger signature of gene conversion among class II than among class I alleles. In Tympanuchus and Centrocercus, 91% and 67% of class II alleles, respectively, were involved in at least one intra-generic gene conversion event. By contrast, 13% of Tympanuchus class I alleles and no Centrocercus class I alleles had signatures of gene conversion. We also found a negative correlation between LD and physical distance between sites at MHC class II, but not at class I, indicating that recombination is important for increasing diversity of class II (Kiemnec-Tyburczy et al., 2012). These contrasting patterns of gene conversion at class I and II might be explained by the presence of gene duplication at class II. Our pyrosequencing results supported the presence of two class II loci in both Centrocercus and Tympanuchus grouse. As gene conversion may occur not only between alleles from the same locus, but also between loci (Ohta, 1999), recombination rate should increase with the number of duplicated loci, although it is expected to be inversely proportional to the distance between loci (Ezawa et al., 2006). While the unlinked MHC loci, such as B and Y, are less prone to inter-locus recombination (Burri et al., 2008), both class II B loci in grouse are separated by less than 7 kb (Wang et al., 2012; Eimes et al., 2013), which should facilitate a high gene conversion rate. Although the evidence for inter-locus recombination at MHC is now well-established in different vertebrate taxa (Hughes and Nei, 1989; Cardenas et al., 2005; Zagalska-Neubauer et al., 2010), including galliforms (Strand et al., 2013), the relative rates of inter- and intra-locus gene conversion at MHC are difficult to assess. The scarce available information suggests that both types of recombination may occur at a similar rate within class II B (Reusch and Langefors, 2005). In addition, high similarity among sequences derived from different MHC loci (apparent paralogs) suggests that inter-locus gene conversion may be an important evolutionary mechanism in fish (Reusch and Langefors, 2005; Aguilar and Garza, 2007) and birds (Edwards et al., 1995; Wittzell et al., 1999a, b; Burri et al., 2008). In grouse, we found that ca. 25% of class II alleles were shared between BLB1 and BLB2, indicating significant homogenisation of both loci. Our evidence for signatures of recombination between Tympanuchus and Centrocercus alleles suggests that class II loci may have been homogenized by inter-locus gene conversion in their ancestor and retained after species diversification. This appears to be rare empirical evidence for the concerted evolution of duplicated MHC genes, often invoked to explain why avian phylogenies based on MHC usually sort by species rather than by locus (Hess and Edwards, 2002).
We also found much more frequent intra-locus gene conversion at the BLB1 class II locus than at the class I locus, as all Tympanuchus BLB1-specific alleles (n=17) were involved in gene conversions events compared with only four of 32 class I alleles. This higher rate of intra-locus gene conversion at class II could be due to stronger selective pressure acting on these genes, or it could be due to an interaction of gene conversion and selection. When gene conversion occurs, even if the rate is low, weak selection per amino acid site is effective for enhancing polymorphisms and may easily increase the number of MHC alleles in the population (Ohta, 1999). Thus, stronger balancing selection might favour new alleles produced by gene conversion leading to greater allelic polymorphism across evolutionary time. Indeed, a meta-analysis of teleost fish found that the dN/dS ratio was positively related to recombination rate (Wegner, 2008). Wegner (2008) suggested that recombination could increase the efficiency of natural selection by eliminating LD between selected codons or combining them from different alleles. Our findings in grouse agree with this hypothesis as we found that higher rates of both balancing selection and gene conversion were associated with higher diversity at class II than class I.
Contrasting evolutionary histories of MHC class I and II genes in grouse were also revealed by their contrasting phylogenies. We found that class I alleles clustered strongly by genus, whereas Centrocerucus and Tympanuchus class II alleles did not form monophyletic clades. There are two non-exclusive mechanisms that may account for these differences. First, we found a strong signal of gene conversion between private Centrocercus and Tympanuchus class II alleles and the signature of inter-generic conversion events was less frequent for class I alleles. As increased recombination decreases phylogenetic signal, Centrocercus and Tympanuchus class II alleles generated by ancestral gene conversion were more likely to cluster together rather than by genus. Secondly, this pattern could be enhanced by the higher intensity of balancing selection acting on class II genes. Under strong balancing selection, MHC alleles are preserved and can persist beyond speciation events (Klein et al., 1993). Such trans-species polymorphism seems to be a general feature of MHC evolution and has been demonstrated in many vertebrate systems (Ottová et al., 2005; Cutrera and Lacey, 2007; Kamath and Getz, 2011), although less often in birds (Hess and Edwards, 2002).
It has to be kept in mind that sharing of identical MHC allelic variants between species may also imply recent speciation events or interspecific hybridisation (Nadachowska-Brzyska et al., 2012). Sharing of MHC alleles between species from the same genus has been found in several recently diverged taxonomic groups, such as Xenopus frogs (Bos and Waldman, 2006), Spheniscus penguins (Bollmer et al., 2007; Kikkawa et al., 2009), equids (Kamath and Getz, 2011) and primates (Doxiadis et al., 2006). The divergence between all three Tympanuchus species likely occurred within the last 0.5–2 million years, and in areas of geographic overlap, hybridisation has been observed between T. cupido and two other Tympanuchus grouse (Galla and Johnson, 2015). Centrocercus minimus is a recently described species and probably at an intermediate stage of speciation from C. urophasianus, driven mostly by reproductive isolation (Young et al., 2000; Oyler-McCance et al., 2010). Thus, recent divergence seems to be a reasonable explanation for the trans-species allele sharing within both grouse genera. On the other hand, we also recorded identical class I and class II allelic variants shared between Centrocercus and Tympanuchus. MHC allele sharing across genera has been observed rarely and is usually attributed to trans-species polymorphism resulting from retention of ancestral allelic variants (Go et al., 2002; Meyer-Lucht et al., 2008).
In conclusion, this is one of the first studies clearly demonstrating contrasting roles of selection and gene conversion in the evolution of MHC class I and class II in wild birds. Our results suggest that macroevolutionary mechanisms can act differently on genes involved in the immune response against intracellular and extracellular pathogens, but more data are needed to generalize this conclusion. Thus, we encourage comparative research on evolutionary histories of MHC class I and class II in other vertebrate systems, as well as studies comparing the evolution of MHC and other immune genes involved in pathogen recognition.
Data archiving
DNA sequences for the newly described MHC alleles will be deposited in GenBank (Accession nos: KU342536-KU342564). DNA sequences for the previously identified alleles are available in GenBank for MHC class I (Accession nos.: JX237361, JX237363–JX237364, JX237366, JX237368–JX237369. KF466475–KF466476, KF466478–KF466479 and KR779965-KR779979) and MHC class II (Accession nos.: FJ232512–FJ232514, FJ232516–FJ232518, GQ176848–GQ176851, HM011573–HM011577, HM011580 and KR779980-KR779993). MHC class I and class II genotypes used in the analyses are in Dryad: doi: 10.5061/dryad.35jt8.
Accession codes
References
Aguilar A, Garza JC . (2007). Patterns of historical balancing selection on the salmonid major histocompatibility complex class II β gene. J Mol Evol 65: 34–43.
Alcaide M, Liu M, Edwards SV . (2013). Major histocompatibility complex class I evolution in songbirds: universal primers, rapid evolution and base compositional shifts in exon 3. PeerJ 1: e86.
Alcaide M, Munoz J, Martínez‐de la Puente J, Soriguer R, Figuerola J . (2014). Extraordinary MHC class II B diversity in a non‐passerine, wild bird: the Eurasian Coot Fulica atra (Aves: Rallidae). Ecol Evol 4: 688–698.
Bateson ZW, Dunn PO, Hull SD, Henschen AE, Johnson JA, Whittingham LA . (2014). Genetic restoration of a threatened population of greater prairie-chickens. Biol Conserv 174: 12–19.
Bellinger MR, Johnson JA, Toepfer J, Dunn P . (2003). Loss of genetic variation in greater prairie chickens following a population bottleneck in Wisconsin, USA. Conserv Biol 17: 717–724.
Bendell JF . (1955). Disease as a control of a population of blue grouse, Dendragapus obscurus fuliginosus (Ridgway). Can J Zool 33: 195–223.
Bernatchez L, Landry C . (2003). MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16: 363–377.
Bollmer JL, Dunn PO, Freeman-Gallant CR, Whittingham LA . (2012). Social and extra-pair mating in relation to major histocompatibility complex variation in common yellowthroats. Proc R Soc B 279: 4778–4785.
Bollmer JL, Dunn PO, Whittingham LA, Wimpee C . (2010). Extensive MHC class II B gene duplication in a passerine, the common yellowthroat (Geothlypis trichas. J Hered 101: 448–460.
Bollmer JL, Vargas FH, Parker PG . (2007). Low MHC variation in the endangered Galapagos penguin (Spheniscus mendiculus. Immunogenetics 59: 593–602.
Bondinas GP, Moustakas AK, Papadopoulos GK . (2007). The spectrum of HLA-DQ and HLA-DR alleles, 2006: a listing correlating sequence and structure with function. Immunogenetics 59: 539–553.
Bonneaud C, Sorci G, Morin V, Westerdahl H, Zoorob R, Wittzell H . (2004). Diversity of Mhc class I and IIB genes in house sparrows (Passer domesticus. Immunogenetics 55: 855–865.
Bos DH, Waldman B . (2006). Evolution by recombination and transspecies polymorphism in the MHC class I gene of Xenopus laevis. Mol Biol Evol 23: 137–143.
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL et al. (1993). Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364: 33–39.
Burri R, Hirzel HN, Salamin N, Roulin A, Fumagalli L . (2008). Evolutionary patterns of MHC class II B in owls and their implications for the understanding of avian MHC evolution. Mol Biol Evol 25: 1180–1191.
Cardenas PP, Suarez CF, Martinez P, Patarroyo ME, Patarroyo MA . (2005). MHC class I genes in the owl monkey: mosaic organisation, convergence and loci diversity. Immunogenetics 56: 818–832.
Chaves LD, Faile GM, Krueth SB, Hendrickson JA, Reed KM . (2010). Haplotype variation, recombination, and gene conversion within the turkey MHC-B locus. Immunogenetics 62: 465–477.
Christiansen TJ, Tate CM . (2011). Parasites and infectious diseases of Greater Sage-Grouse. In: Knick S, Connelly JW (eds) Greater Sage-Grouse: Ecology and Conservation of a Landscape Species and its Habitats. University of California Press: Berkeley, CA, USA. pp 113–126.
Clarke B, Kirby DRS . (1966). Maintenance of histocompatibility polymorphisms. Nature 211: 999–1000.
Cullen M, Perfetto SP, Klitz W, Nelson G, Carrington M . (2002). High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am J Hum Genet 71: 759–776.
Cutrera AP, Lacey EA . (2007). Trans-species polymorphism and evidence of selection on class II MHC loci in tuco-tucos (Rodentia: Ctenomyidae). Immunogenetics 59: 937–948.
Delport W, Poon AF, Frost SD, Pond SLK . (2010). Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26: 2455–2457.
Doxiadis GG, Rouweler AJ, de Groot NG, Louwerse A, Otting N, Verschoor EJ et al. (2006). Extensive sharing of MHC class II alleles between rhesus and cynomolgus macaques. Immunogenetics 58: 259–268.
Drew ML, Wingle WL, Graham DL, Griffin CP, Silvy NJ, Fadly AM et al. (1998). Reticuloendotheliosis in captive greater and Attwater’s prairie chickens. J Wildlife Dis 34: 783–791.
Drouin G, Prat F, Ell M, Clarke GD . (1999). Detecting and characterizing gene conversions between multigene family members. Mol Biol Evol 16: 1369–1390.
Edwards SV, Gasper J, March M . (1998). Genomics and polymorphism of Agph-DAB1, an Mhc class II B gene in red-winged blackbirds (Agelaius phoeniceus. Mol Biol Evol 15: 236–250.
Edwards SV, Wakeland EK, Potts WK . (1995). Contrasting histories of avian and mammalian Mhc genes revealed by class II B sequences from songbirds. Proc Natl Acad Sci USA 92: 12200–12204.
Eimes JA, Bollmer JL, Dunn PO, Whittingham LA, Wimpee C . (2010). Mhc class II diversity and balancing selection in greater prairie-chickens. Genetica 138: 265–271.
Eimes JA, Bollmer JL, Whittingham LA, Johnson JA, van Oosterhout C, Dunn PO . (2011). Rapid loss of MHC class II variation in a bottlenecked population is explained by drift and loss of copy number variation. J Evol Biol 24: 1847–1856.
Eimes JA, Reed KM, Mendoza KM, Bollmer JL, Whittingham LA, Bateson ZW et al. (2013). Greater prairie-chickens have a compact MHC-B with a single class IA locus. Immunogenetics 65: 133–144.
Ejsmond MJ, Radwan J, Wilson AB . (2014). Sexual selection and the evolutionary dynamics of the major histocompatibility complex. Proc R Soc B 281: 20141662.
Ezawa K, OOta S, Saitou N . (2006). Genome-wide search of gene conversions in duplicated genes of mouse and rat. Mol Biol Evol 23: 927–940.
Galan M, Guivier E, Caraux G, Charbonnel N, Cosson JF . (2010). A 454 multiplex sequencing method for rapid and reliable genotyping of highly polymorphic genes in large-scale studies. BMC Genomics 11: 296.
Galla SJ, Johnson JA . (2015). Differential introgression and effective size of marker type influence phylogenetic inference of a recently divergent avian group (Phasianidae: Tympanuchus. Mol Phylogenet Evol 84: 1–13.
Galtier N, Piganeau G, Mouchiroud D, Duret L . (2001). GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159: 907–911.
Gangoso L, Alcaide M, Grande JM, Muñoz J, Talbot SL, Sonsthagen SA et al. (2012). Colonizing the world in spite of reduced MHC variation. J Evol Biol 25: 1438–1447.
Garrigan D, Hedrick PW . (2003). Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution 57: 1707–1722.
Go Y, Satta Y, Kawamoto Y, Rakotoarisoa G, Randrianjafy A, Koyama N et al. (2002). Mhc-DRB genes evolution in lemurs. Immunogenetics 54: 403–417.
Gross AO . (1925). Diseases of the ruffed grouse. Science 62: 55–57.
Gross AO . (1928). The heath hen. Mem Boston Soc Nat Hist 6: 490–588.
Harper GR, Klataske RD, Robel RJ, Hansen MF . (1967). Helminths of greater prairie chickens in Kansas. J Wildlife Manage 31: 265–269.
Hedrick PW . (2002). Pathogen resistance and genetic variation at MHC loci. Evolution 56: 1902–1908.
Hess CM, Edwards SV . (2002). The evolution of the major histocompatibility complex in birds. Bioscience 52: 423–431.
Hill WG, Robertson A . (1968). Linkage disequilibrium in finite populations. Theor Appl Genet 38: 226–231.
Hosomichi K, Miller MM, Goto RM, Wang Y, Suzuki S, Kulski JK et al. (2008). Contribution of mutation, recombination, and gene conversion to chicken MHC-B haplotype diversity. J Immunol 181: 3393–3399.
Hudson PJ, Newborn D, Dobson AP . (1992). Regulation and stability of a free-living host-parasite system: Trichostrongylus tenuis in red grouse. I. Monitoring and parasite reduction experiments. J Anim Ecol 61: 477–486.
Hudson RR, Kaplan NL . (1985). Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164.
Hughes AL . (1999) Adaptive Evolution of Genes and Genomes. Oxford University Press: New York, NY, USA.
Hughes AL, Nei M . (1988). Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335: 167–170.
Hughes AL, Nei M . (1989). Ancient interlocus exon exchange in the history of the HLA-A locus. Genetics 122: 681–686.
Janeway C, Travers P, Walport M, Shlomchick M . (2005) Immunobiology: the Immune System in Health and Disease. Garland Press: New York, NY, USA.
Jarvi SI, Tarr CL, Mcintosh CE, Atkinson CT, Fleischer RC . (2004). Natural selection of the major histocompatibility complex (Mhc) in Hawaiian honeycreepers (Drepanidinae). Mol Ecol 13: 2157–2168.
Jukes TH, Cantor CR . (1969). Evolution of protein molecules. In: Munro HN (ed). Mammalian Protein Metabolism. Academic Press: New York, NY, USA. pp 21–132.
Kamath PL, Getz WM . (2011). Adaptive molecular evolution of the Major Histocompatibility Complex genes, DRA and DQA, in the genus Equus. BMC Evol Biol 11: 128.
Kaufman J . (1999). Co-evolving genes in MHC haplotypes: the ‘rule’ for nonmammalian vertebrates? Immunogenetics 50: 228–236.
Kaufman J, Andersen R, Avila D, Engberg J, Lambris J, Salomonsen J et al. (1992). Different features of the MHC class I heterodimer have evolved at different rates. Chicken BF and beta 2-microglobulin sequences reveal invariant surface residues. J Immunol 148: 1532–1546.
Kaufman J, Milne S, Göbel TW, Walker BA, Jacob JP, Auffray C et al. (1999). The chicken B locus is a minimal essential major histocompatibility complex. Nature 401: 923–925.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649.
Kelley J, Walter L, Trowsdale J . (2005). Comparative genomics of major histocompatibility complexes. Immunogenetics 56: 683–695.
Kiemnec-Tyburczy KM, Richmond JQ, Savage AE, Lips KR, Zamudio KR . (2012). Genetic diversity of MHC class I loci in six non-model frogs is shaped by positive selection and gene duplication. Heredity 109: 146–155.
Kikkawa EF, Tsuda TT, Sumiyama D, Naruse TK, Fukuda M, Kurita M et al. (2009). Trans-species polymorphism of the Mhc class II DRB-like gene in banded penguins (genus Spheniscus. Immunogenetics 61: 341–352.
Klein J, Sato A, Nagel S, O’hUigín C . (1998). Molecular trans-species polymorphism. Annu Rev Ecol Syst 29: 1–21.
Klein J, Satta Y, Takahata N, O’hUigin C . (1993). Trans-specific Mhc polymorphism and the origin of species in primates. J Med Primatol 22: 57–64.
Klitz W, Thomson G, Baur MP . (1986). Contrasting evolutionary histories among tightly linked HLA loci. Am J Hum Genet 39: 340–349.
Kosakovsky Pond SLK, Frost SD . (2005). Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22: 1208–1222.
Kruiswijk CP, Hermsen T, van Heerwaarden J, Dixon B, Savelkoul HF, Stet RJ . (2005). Major histocompatibility genes in the Lake Tana African large barb species flock: evidence for complete partitioning of class II B, but not class I, genes among different species. Immunogenetics 56: 894–908.
Kuduk K, Babik W, Bojarska K, Śliwińska EB, Kindberg J, Taberlet P et al. (2012). Evolution of major histocompatibility complex class I and class II genes in the brown bear. BMC Evol Biol 12: 197.
Leigh WH . (1941). Variation in a new species of cestode, Raillietina Skrjabinia variabila, from the prairie-chicken in Illinois. J Parasitol 27: 97–106.
Lenz TL, Becker S . (2008). Simple approach to reduce PCR artefact formation leads to reliable genotyping of MHC and other highly polymorphic loci—implications for evolutionary analysis. Gene 427: 117–123.
Lewontin RC . (1964). The interaction of selection and linkage. I. Genetic considerations; heterotic models. Genetics 49: 49–67.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Lighten J, van Oosterhout C, Paterson IG, McMullan M, Bentzen P . (2014). Ultra‐deep Illumina sequencing accurately identifies MHC class IIb alleles and provides evidence for copy number variation in the guppy (Poecilia reticulata. Mol Ecol Res 14: 753–767.
Mansai SP, Innan H . (2010). The power of the methods for detecting interlocus gene conversion. Genetics 184: 517–527.
Meunier J, Eyre-Walker A . (2001). The correlation between linkage disequilibrium and distance: implications for recombination in hominid mitochondria. Mol Biol Evol 18: 2132–2135.
Meyer-Lucht Y, Otten C, Püttker T, Sommer S . (2008). Selection, diversity and evolutionary patterns of the MHC class II DAB in free-ranging Neotropical marsupials. BMC Genet 9: 39.
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL . (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8: e1002764.
Nadachowska-Brzyska K, Zieliński P, Radwan J, Babik W . (2012). Interspecific hybridization increases MHC class II diversity in two sister species of newts. Mol Ecol 21: 887–906.
Nei M, Gojobori T . (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426.
Nei M, Gu X, Sitnikova T . (1997). Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci USA 94: 7799–7806.
Nei M, Rooney AP . (2005). Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39: 121–152.
Ohta T . (1999). Effect of gene conversion on polymorphic patterns at major histocompatibility complex loci. Immunol Rev 167: 319–325.
Otting N, Heijmans CM, Noort RC, de Groot NG, Doxiadis GGM, van Rood JJ et al. (2005). Unparalleled complexity of the MHC class I region in rhesus macaques. Proc Natl Acad Sci USA 102: 1626–1631.
Ottová E, Šimková A, Martin JF, De Bellocq JG, Gelnar M, Allienne JF et al. (2005). Evolution and trans-species polymorphism of MHC class IIβ genes in cyprinid fish. Fish Shellfish Immunol 18: 199–222.
Oyler-McCance SJ, St John J, Quinn TW . (2010). Rapid evolution in lekking grouse. Ornithol Monogr 67: 114–122.
Oyler-McCance SJ, St John J, Taylor SE, Apa AD, Quinn TW . (2005). Population genetics of Gunnison Sage-Grouse: Implications for management. J Wildl Manage 69: 630–637.
Peterson MJ . (2004). Parasite and infectious diseases of prairie grouse: should managers be concerned? Wildlife Soc Bull 32: 35–55.
Peterson MJ, Ferro PJ, Peterson MN, Sullivan R, Toole BE, Silvy NJ . (2002). Infectious disease survey of lesser prairie chickens in the northern Rolling Plains of Texas. J Wildlife Dis 38: 834–839.
Posada D . (2002). Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol Biol Evol 19: 708–717.
Promerová M, Králová T, Bryjová A, Albrecht T, Bryja J . (2013). MHC class IIB exon 2 polymorphism in the grey partridge (Perdix perdix is shaped by selection, recombination and gene conversion. PLoS One 8: e69135.
Radwan J, Zagalska-Neubauer M, Cichoń M, Sendecka J, Kulma K, Gustafsson L et al. (2012). MHC diversity, malaria and lifetime reproductive success in collared flycatchers. Mol Ecol 21: 2469–2479.
Reusch TB, Langefors Å . (2005). Inter-and intralocus recombination drive MHC class IIB gene diversification in a teleost, the three-spined stickleback Gasterosteus aculeatus. J Mol Evol 61: 531–541.
Robinson J, Halliwell JA, McWilliam H, Lopez R, Parham P, Marsh SG . (2013). The IMGT/HLA database. Nucleic Acids Res 41: D1222–D1227.
Sawyer S . (1989). Statistical tests for detecting gene conversion. Mol Biol Evol 6: 526–538.
Schaschl H, Wandeler P, Suchentrunk F, Obexer-Ruff G, Goodman SJ . (2006). Selection and recombination drive the evolution of MHC class II DRB diversity in ungulates. Heredity 97: 427–437.
Schaschl H, Wegner KM . (2007). Contrasting mode of evolution between the MHC class I genomic region and class II region in the three-spined stickleback (Gasterosteus aculeatus L.; Gasterosteidae: Teleostei). Immunogenetics 59: 295–304.
Scheffler K, Martin DP, Seoighe C . (2006). Robust inference of positive selection from recombining coding sequences. Bioinformatics 22: 2493–2499.
Sepil I, Moghadam HK, Huchard E, Sheldon BC . (2012). Characterization and 454 pyrosequencing of Major Histocompatibility Complex class I genes in the great tit reveal complexity in a passerine system. BMC Evol Biol 12: 68.
Shaw JL . (1990). Effects of the caecal nematode Trichostrongylus tenuis on egg-laying by captive red grouse. Res Vet Sci 48: 59–63.
Shum BP, Guethlein L, Flodin LR, Adkison MA, Hedrick RP, Nehring RB et al. (2001). Modes of salmonid MHC class I and II evolution differ from the primate paradigm. J Immunol 166: 3297–3308.
Siddle HV, Sanderson C, Belov K . (2007). Characterization of major histocompatibility complex class I and class II genes from the Tasmanian devil (Sarcophilus harrisii. Immunogenetics 59: 753–760.
Spurgin LG, Richardson DS . (2010). How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B 277: 979–988.
Spurgin LG, van Oosterhout C, Illera JC, Bridgett S, Gharbi K, Emerson BC et al. (2011). Gene conversion rapidly generates major histocompatibility complex diversity in recently founded bird populations. Mol Ecol 20: 5213–5225.
Stein RW, Brown JW, Mooers AØ . (2015). A molecular genetic time scale demonstrates Cretaceous origins and multiple diversification rate shifts within the order Galliformes (Aves). Mol Phylogent Evol 95: 155–164.
Strand T, Wang B, Meyer-Lucht Y, Höglund J . (2013). Evolutionary history of black grouse major histocompatibility complex class IIB genes revealed through single locus sequence-based genotyping. BMC Genet 14: 29.
Strand T, Westerdahl H, Hoglund J, Alatalo RV, Siitari H . (2007). The Mhc class II of the Black grouse (Tetrao tetrix consists of low numbers of B and Y genes with variable diversity and expression. Immunogenetics 59: 725–734.
Strandh M, Lannefors M, Bonadonna F, Westerdahl H . (2011). Characterization of MHC class I and II genes in a subantarctic seabird, the blue petrel, Halobaena caerulea (Procellariiformes). Immunogenetics 63: 653–666.
Stuglik MT, Radwan J, Babik W . (2011). jMHC: software assistant for multilocus genotyping of gene families using next-generation amplicon sequencing. Mol Ecol Res 11: 739–742.
Takahata N, Nei M . (1990). Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics 124: 967–978.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S . (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30: 2725–2729.
Wang B, Ekblom R, Strand TM, Portela-Bens S, Höglund J . (2012). Sequencing of the core MHC region of black grouse (Tetrao tetrix and comparative genomics of the galliform MHC. BMC Genomics 13: 553.
Wegner KM . (2008). Historical and contemporary selection of teleost MHC genes: did we leave the past behind? J Fish Biol 73: 2110–2132.
Winternitz JC, Wares JP . (2013). Duplication and population dynamics shape historic patterns of selection and genetic variation at the major histocompatibility complex in rodents. Ecol Evol 3: 1552–1568.
Wittzell H, Bernot A, Auffray C, Zoorob R . (1999a). Concerted evolution of two Mhc class II B loci in pheasants and domestic chickens. Mol Biol Evol 16: 479–490.
Wittzell H, Madsen T, Westerdahl H, Shine R, von Schantz T . (1999b). MHC variation in birds and reptiles. Genetica 104: 301–309.
Young JR, Braun CE, Oyler-McCance SJ, Hupp JW, Quinn TW . (2000). A new species of sage-grouse (Phasianidae: Centrocercus from southwestern Colorado. Wilson Bull 112: 445–453.
Zagalska-Neubauer M, Babik W, Stuglik M, Gustafsson L, Cichoń M, Radwan J . (2010). 454 sequencing reveals extreme complexity of the class II major histocompatibility complex in the collared flycatcher. BMC Evol Biol 10: 395.
Zhang J, Rosenberg HF, Nei M . (1998). Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA 95: 3708–3713.
Acknowledgements
Funding was provided by grants from the Research Growth Initiative, University of Wisconsin-Milwaukee Graduate School, and National Science Foundation (DEB-0948695) to POD and LAW and a Fulbright Fellowship to PM. We thank A Spaulding, D Wolfe and J Toepfer for tissue collection. Any mention of trade, product or firm names is for descriptive purposes only and does not imply endorsement by the US Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Minias, P., Bateson, Z., Whittingham, L. et al. Contrasting evolutionary histories of MHC class I and class II loci in grouse—effects of selection and gene conversion. Heredity 116, 466–476 (2016). https://doi.org/10.1038/hdy.2016.6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2016.6
This article is cited by
-
Distinct evolutionary trajectories of MHC class I and class II genes in Old World finches and buntings
Heredity (2021)
-
Polymorphism and varying selection within the MHC class I of four Anas species
Immunogenetics (2021)
-
Allelic diversity and patterns of selection at the major histocompatibility complex class I and II loci in a threatened shorebird, the Snowy Plover (Charadrius nivosus)
BMC Evolutionary Biology (2020)
-
Allelic diversity and selection at the MHC class I and class II in a bottlenecked bird of prey, the White-tailed Eagle
BMC Evolutionary Biology (2019)
-
Extensive shared polymorphism at non-MHC immune genes in recently diverged North American prairie grouse
Immunogenetics (2018)
