
Abstract
Social and cultural factors had a critical role in determining the genetic structure of Europe. Therefore, socially stratified populations may help to focus on specific episodes of European demographic history. In this study, we use uniparental markers to analyse the genetic structure of Partecipanza in San Giovanni in Persiceto (Northern Italy), a peculiar institution whose origins date back to the Middle Ages and whose members form the patrilineal descent of a group of founder families. From a maternal point of view (mtDNA), Partecipanza is genetically homogeneous with the rest of the population. However, we observed a significant differentiation for Y-chromosomes. In addition, by comparing 17 Y-STR profiles with deep-rooted paternal pedigrees, we estimated a Y-STR mutation rate equal to 3.90 * 10−3 mutations per STR per generation and an average generation duration time of 33.38 years. When we used these values for tentative dating, we estimated 1300-600 years ago for the origins of the Partecipanza. These results, together with a peculiar Y-chromosomal composition and historical evidence, suggest that Germanic populations (Lombards in particular) settled in the area during the Migration Period (400–800 AD, approximately) and may have had an important role in the foundation of this community.
Similar content being viewed by others

Introduction
Recent demographic episodes and population events are rarely studied by geneticists and molecular anthropologists. In fact, if human genetic diversity can be described as a palimpsest in which different layers—reflecting different episodes—accumulate through time, dissecting a single event is a difficult task. Furthermore, recent events are rarely associated with the substitution of large populations. On the other hand, such events just partially modify the pre-existing genetic background, hence their genetic ‘signals’ are generally low and difficult to detect. These are some of the reasons why many studies focus on some well-known events and try to link them to observed genetic patterns (Jobling, 2012; Larmuseau et al., 2013). Ancient DNA research is only beginning to discover what stands behind elusive labels such as ‘Neolithic’, ‘Linear Pottery Culture’, ‘Etruscans’ and so on, but the actual genetic variability of ancient populations still remains largely unknown (Lacan et al., 2012).
It has been recently shown that an adequate sampling strategy may help to address some of these problems. For instance, ‘marginal’ populations, such as ethno-linguistic minorities or geographic isolates, not only give an important contribution to the biodiversity of a region, but are more likely to display genetic features that were masked by intervening demographic episodes in open populations (Boattini et al., 2011a, 2011b; Capocasa et al., 2014). As a second point, surnames and/or pedigrees can be used for selecting samples within populations. Sampling males bearing surnames that are unequivocally associated with a certain place for Y-chromosome studies provides proxies for older populations (King and Jobling, 2009a; Boattini et al., 2012). The same considerations hold for sample selection based on genealogies/pedigrees. Compared to surnames, genealogies yield a higher degree of temporal and geographic precision. Extensive genealogical data, on the other hand, are rare and their collection is considerably time-consuming. A small but increasing number of studies has shown that surname- and pedigree-based samplings can reveal otherwise hidden genetic structures (Larmuseau et al., 2013 and references therein).
As recently observed (Boattini et al., 2013), the genetic history of Italy is particularly complicated in comparison with other European countries. We focussed on a particularly interesting population, Partecipanza of San Giovanni in Persiceto, so that some of its less-known aspects can be better understood. From a juridical point of view, Partecipanze are an absolutely idiosyncratic way of sharing and devolving collective lands. These institutions originated in the Middle Ages and are still present in some areas of Northern Italy. The privilege to participate (i.e., to share the leased assets) is inherited following a gene-like pattern through exclusive admission granted to the descent of a group of founder families, usually following the paternal line (Zanarelli, 1992). In this way, the members of Partecipanza conserved through the centuries their social and economic identity, potentially together with some of their genetic features. Furthermore, thanks to the wide archival documentation conserved by this institution, it is possible to compare DNA samples with paternal pedigrees for the last four to five centuries. All these features make the Partecipanza an exceptional observatory on the recent genetic history of Italy.
In this study, we explore the paternal (Y-chromosome) and maternal (mtDNA) genetic variability of Partecipanza of San Giovanni in Persiceto (PAR). Results are compared with those of a set of ‘control’ individuals sampled in the same place, but not sharing the affiliation to Partecipanza (SGP), and interpreted considering a wide set of reference populations from Italy and Europe. Our main aims are: (i) to check the genetic effects—if any—of the social-economical separation between PAR and SGP; (ii) to reconstruct the time and the genetic origins of the Partecipanza, as well as their implications for the genetic history of Italy; and (iii) to estimate Y-STR mutation rates and average generation duration time by comparing paternal pedigrees and Y-chromosomal haplotypes.
Materials and methods
The population
Partecipanze are located in the flat portions of the provinces of Bologna and Ferrara (Padana plain, Northern Italy). Their origins are related to medieval events of land reclamation, but the exact date of their foundation is unknown. Evolving from ancient collective emphyteutic grants, Partecipanze soon became reserved to a restricted group of founder families. Currently, six Partecipanze are still present and active in the study area: San Giovanni in Persiceto, Nonantola, Cento, Pieve di Cento, Sant'Agata Bolognese and Villa Fontana. Among these, Partecipanza of San Giovanni in Persiceto is probably the most important. Its shared assets exceed ∼2400 ha and its population size is of ∼5000 individuals. This Partecipanza (to which we will refer exclusively from now on) is historically documented from at least 1170 and 1215 AD, when emphyteutic grants to the community of S. Giovanni in Persiceto (respectively, from the abbot of Nonantola and the bishop of Bologna) were stipulated. The customary laws that regulated the affiliation to the Partecipanza were definitively coded around the year 1500 AD (capitula). According to these agreements, the privilege to participate (i.e., to be a member of Partecipanza) was conditioned by two main rules: being a patrilineal legitimate descent from a Partecipante family and to maintain the place of residence within the legal boundaries of S. Giovanni in Persiceto. In addition, the procedure of cavazione was established. Cavazione is a periodic re-shuffling of shared lands among heads of the participating households. This procedure is usually repeated each 9 years, leaving us census-like descriptions of the population that are conserved in the archive of the institution. Thanks to their favoured access to land, members of Partecipanza for centuries constituted the élite of San Giovanni in Persiceto. Such dominance was highlighted by the fact that the leading council of Partecipanza was at the same time the head of the local Commune. Only in 1833, the two institutions (Partecipanza and Commune) were definitively separated. At present, the individuals/families affiliated to the Partecipanza are characterised by 38 different surnames (Zanarelli, 1992). The exact dates of origin of each Partecipanza surname are not currently determined, because of the scarcity of archival data before the seventeenth century. However, according to what is known for Northern Italy and in particular for rural areas, it is reasonable to hypothesize that most of these family names spread during the sixteenth century (Boattini et al., 2012 and bibliography therein).
DNA samples
Buccal swabs were collected from 149 male individuals sampled in San Giovanni in Persiceto and its surroundings. Among them, 88 belong to the Partecipanza (PAR sample), while the remaining 61 form an ‘open’ sample (SGP sample), sharing the same environmental and cultural features, aside from the status of Partecipante. All these samples were collected according to the standard ‘grandparents’ criterion (i.e., at least three generations of ancestry in the SGP area) and excluding related individuals (up to second cousins). To best reconstruct the paternal genetic variability of the Partecipanza, we sampled at least one individual for each of the 38 Partecipanza surnames. As for the SGP sample, all the selected individuals share surnames that were identified as autochthonous for the considered area by Boattini et al. (2012). To avoid any surname-related bias, all SGP individuals bear different surnames (with the negligible exception of three surnames that are represented by two individuals each). Data from 14 individuals have been previously published in Boattini et al. (2013).
The collection of biological samples was performed during various sessions from 2008 to 2012. For all subjects, a written informed consent was obtained and the Ethics Committees at the Azienda Ospedaliero-Universitaria Policlinico S.Orsola-Malpighi of Bologna (Italy) approved all procedures. The confidentiality of personal information for each participant to the study was assured.
Comparison populations
Reference data include 32 populations from: North-Western Italy, South-Eastern Italy and Sicily (21, Boattini et al., 2013; Sarno et al., 2014), French Basques (3, Martínez-Cruz et al., 2012), Germany (3, Rębała et al., 2013), Poland and Slovakia (4, Rębała et al., 2013), Balkans (1, Regueiro et al., 2012). A full list is available in Supplementary Table S1 and their geographic position is represented in Supplementary Figure S1. In addition, four haplotypes for Y-chromosome hg I1-P109 from Brabant (Larmuseau et al., 2011; Larmuseau, personal communication) were considered.
Y-chromosome genotyping
PCR amplification of 17 Y-STR loci (DYS19,DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS385a/b, DYS437, DYS438, DYS439, DYS448, DYS456, DYS458, DYS635 and GATA H4) was carried out by using the AmpFlSTR Yfiler PCR Amplification Kit (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s recommendations in a final volume of 5 μl. The PCR reaction consisted of denaturation at 95 °C for 11 min, followed by 30 denaturation cycles at 94 °C for 1 min, annealing at 61 °C for 1 min, extension at 72 °C for 1 min and a final extension at 60 °C for 80 min. Products were sized on an ABI Prism 310 Genetic Analyzer using the GeneScan 3.7 software (Applied Biosystems). As the Yfiler kit amplifies DYS385a/b simultaneously, avoiding the determination of each of the two alleles (a or b), these loci were excluded from downstream analyses except where explicitly specified. DYS389b was obtained by subtracting DYS389I from DYS389II. Basal haplogroups (hgs) were assigned by typing the seven SNPs (R-M173, J-M172, I-M170, E-M35, K-M9, P-M45 and F-M89) implemented in the MY1 Multiplex PCR by Onofri et al. (2006). Subsequently, we explored Y-chromosome variability by using 28 SNPs. Twenty-six of them (E-M78, E-V12, E-V13, E-V22, G-P15, G-P16, G-M286, G-U8, G-U13, I-M253, I-M227, I-L22, I-P215, I-M26, J-M410, J-L27, J-M67, J-M92, J-M12, R-M17, R-M343, R-M18, R-M269, R-S21/U106, R-SRY2627/M167 and R-S28/U152) were included in five hg-specific multiplexes (Ferri and Alù, 2012). The SNP genotyping was carried out by means of PCR Multiplex amplification, followed by Minisequencing reaction based on dideoxy Single Base Extension, which was performed with the SNaPshot multiplex kit (Applied Biosystems). Single Base Extension products were analysed with capillary electrophoresis on an ABI Prism 310 Genetic Analyser. Two additional SNPs (E-M81, E-M123) were finally tested with RFLP analysis, by using HpyCH4IV and DdeI enzymes, respectively, as in Sarno et al. (2014).
Hgs nomenclature is in accordance with the Y-Chromosome Consortium (Karafet et al., 2008, later updated by van Oven et al., 2014). For reasons of simplicity, hgs were referred on the base of their defining SNPs. Individual hg information and Y-STR data are provided in Supplementary Table S2.
Mitochondrial DNA genotyping
Variation at the mtDNA HVS-I and HVS-II regions was investigated by sequencing a total of 750 base pairs encompassing nucleotide positions from 15975 to 155. PCR of the HVSI/II regions was carried out in a T-Gradient Thermocycler (Whatman Biometra, Gottingen, Germany) with the following amplification profile: initial denaturation 95 °C for 5 min, 35 cycles of 95 °C for 30 s, 58 °C for 30 s, 72 °C for 5 min and final extension at 72 °C for 15 min. PCR products were purified by ExoSap-IT1 (USB Corporation, Cleveland, OH, USA) and sequenced on an ABI Prism 3730 Genetic Analyzer (Applied Biosystems) by using a Big-Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) according to the manufacturer’s instructions. To reduce ambiguities in sequence determination, the forward and reverse primers were used to sequence both strands of HVS-I and HVS-II regions. The CHROMAS 2.33 software was used to read the obtained electropherograms. Sequences were finally aligned to both the Revised Cambridge reference sequence (Andrews et al., 1999) and the new Reconstructed Sapiens Reference Sequence (Behar et al., 2012) by using the DNA Alignment Software 1.3.1.1 (http://www.fluxusengineering.com/align.htm). Sequence data are provided in Supplementary Table S3.
Archival data and pedigree reconstruction
The Historic Archive of Consorzio dei Partecipanti kept a record of enrolments (Registri delle Iscrizioni) since the early seventeenth century. The right to participate to the sharing of leased assets (i.e., to be a Partecipante) was (and still is) based on these registers. Each registration includes the name of the head of the household, his age and/or his year of birth, the composition of his family, his parish/locality of residence and all the additional information needed to qualify him as a Partecipante (e.g., the code of the precedent enrolment). These registers were compiled in occasion of cavazione, usually each 9 years.
We examined the whole series of the available Registri delle Iscrizioni, at present composed by registers from the years 1606, 1612, 1631, 1634, 1640, 1643, 1650, 1653, 1659, 1662, 1668, 1671, 1677, 1680, 1686, 1689, 1695, 1698, 1707, 1713, 1716, 1722, 1725, 1731, 1740, 1743, 1752, 1761, 1764, 1770, 1779, 1788, 1797, 1806, 1815, 1824, 1833, 1842, 1851, 1860, 1869, 1878, 1887, 1896, 1905, 1914, 1923, 1932, 1941, 1950, 1959, 1968, 1977, 1986, 1995, 2004.
Information from these registers was used to draw paternal pedigrees for the individuals included in our Partecipanza sample. When two or more individuals were found to share a recent paternal ancestor, they were grouped into a single pedigree.
Generation duration time and Y-STR mutation rates
Average generation time was obtained starting from the ages and/or years of birth of the individuals included in the reconstructed pedigrees. For each pedigree, we calculated the number of years and the number of generations encompassed between its root and leaves. The most remote ancestors were excluded, because their year of birth is uncertain. Finally, we divided the total number of years for the total number of generations. Confidence Intervals (95%) were calculated by randomly sampling branches of the pedigrees with a bootstrap procedure (1000 replications).
Y-STR mutation rates were estimated comparing haplotype information within paternal pedigrees that include at least two leaves. Any individual whose hg is not compatible with the reconstructed pedigrees was excluded from calculations. Outlier haplotypes—that is haplotypes showing an outlier number of mutations from the common ancestor—where excluded. Outlier haplotypes were identified by means of a Grubbs test for one outlier in a data sample (function grubbs test, outliers package, R software: Komsta, 2011). For each pedigree, we computed the number of mutation events according to the maximum parsimony method and the number of generations separating the considered individuals/haplotypes. The average STR mutation rate was calculated by dividing the total number of mutations for the total number of generations and for the number of considered STRs. Bootstrapped confidence intervals (CI, 95%) were calculated by randomly re-sampling mutations per STR (1000 replications). To allow maximal resolution, DYS385 was included in the calculations as two separate loci; however, because mutation events were computed within pedigrees, it is highly improbable that apparently identical DYS385 configurations were actually two different haplotypes. In addition, we calculated historical non-paternity rate using the pedigree-based method proposed by Larmuseau et al. (2014). Because this method considers only pairs of related individuals, pedigrees including more than two individuals were represented by pairs of samples separated by the highest number of generations.
Within- and between-population analyses
Standard within-population diversity parameters (Gene Diversity, Mean Number of Pairwise Differences, Nucleotide Diversity) for Y-chromosome (hgs, STR haplotypes) and mtDNA (HVR-I and II sequences), as well as Mismatch Distribution patterns (mtDNA sequences) were calculated with Arlequin 3.5.1.2 (Excoffier et al., 2007). The same software was used to compute pairwise Fst values between PAR and SGP based on Y-chromosome hgs and mtDNA sequences; P-values (following the null hypothesis of no differentiation) were simulated by means of a permutation procedure (1000 replications). Fisher exact tests were applied to determine if differences in Y-hg frequencies between PAR and SGP were statistically significant.
To check the position of our populations within the Italian and European Y-chromosomal landscape, we performed a Non-metric multi-dimensional scaling (MDS). Because different studies used different levels of hg resolution, the analysis was based on 15 Y-STR haplotypes that were available for all the considered reference populations. Calculations were performed using Reynolds distance (Reynolds et al., 1983; for the use of this distance with STRs see Laval et al., 2002) and the function isoMDS implemented in the R software MASS package (R Core Team, 2013). The first and the second Dimension were represented in a scatterplot, along with the corresponding stress value.
Within Y-hgs analysis
To explore genetic variability within those hgs that mostly differentiate PAR from SGP, we applied a Discriminant Analysis of Principal Components (DAPC: Jombart et al., 2010) to Y-STR haplotypes as in Boattini et al. (2013). This analysis is aimed to (i) identify well-resolved groups of haplotypes within hgs; (ii) highlight possible affinities/similarities with reference haplotypes from Italy and Europe; and (iii) constitute a starting point for time estimates. Briefly, the procedure involves two steps. First, haplotypes are grouped using k-means, a clustering algorithm that finds a given number of clusters maximizing the variation between groups. Second, DAPC is used to describe the diversity (and the degree of separation) among such groups of observations, by maximizing the between-group variance and minimizing the within-group variance. The first two to three Discriminant Functions are represented with scatterplots. All analyses were performed within the R software package adegenet (Jombart, 2008).
Y-chromosome time estimates
Time estimates will focus on well-differentiated groups of haplotypes, as described by DAPC. To avoid sampling biases, only clusters comprising at least 10 individuals are considered. Calculations use two different sets of mutation rates: (i) our pedigree-based average mutation rate and (ii) Ballantyne et al. (2010) STR-specific mutation rates. Ballantyne et al. (2010) rates, being averagely similar to our rate (see Results) but available for single STRs, were introduced for further information. Dates are computed with standard deviation (SD) estimator (Sengupta et al. 2006) as in Boattini et al. (2013). Because population events involving PAR are relatively recent, the biasing effect of STRs saturation through time is negligible and all STRs (minus DYS385a/b) are used for calculations. This method estimates the amount of time needed to evolve the observed STRs variation within the given clusters of haplotypes.
Results
Paternal pedigrees, STR mutation rates and generation duration time
We reconstructed paternal lineages (up to the early seventeenth century) for 74 out of 88 individuals of the PAR sample. The remaining 14 individuals were excluded for they presented ambiguities within their paternal pedigrees (e.g., lineages characterised by the very same forenames), as well as reduced legibility. Because some individuals share a common ancestor, these paternal lineages were grouped in 41 independent paternal pedigrees. Fifteen of these pedigrees (encompassing 48 individuals in total) include two or more sampled individuals, up to a maximum of eight (Table 1, Supplementary Figure S2). All individuals included in a given pedigree share the same surname, but a given surname may be present in more than one pedigree (Supplementary Table S2). This could happen because the most recent common ancestor was not reached by documented pedigrees, or because the surname is polyphyletic. Y-STR mutation rates were estimated based on these 15 paternal pedigrees.
As a preliminary step, we excluded from calculations four individuals displaying a different hg compared with the other members of their respective pedigrees and one individual displaying five mutations from the common ancestor of his pedigree. This figure is significantly higher than all the other cases considered here (Supplementary Table S2) and clearly constitutes a case of outlier (Gibbs outlier test: G=4.1066, U=0.5987, P-value=8.63 * 10−5). Finally, we observed a three-step mutation case at locus DYS439 within pedigree P14 (Table 1, Supplementary Table S2). Because it is highly improbable that three independent mutation events involved the very same locus (in a relatively short amount of time), we consider it as a single multi-step mutational event.
By considering 43 individuals across 15 pedigrees and 17 Y-STRs, we observed 24 mutations within 362 generations (Table 1). These values give an average mutation rate equal to 3.90 * 10−3 (95% CI: 2.44 * 10−3, 5.68 * 10−3). Historical non-paternity rate resulted in an estimate of 1.21% (95% CI: 0.4%, 6.13%) per generation.
Average generation duration time was calculated using all the 31 paternal pedigrees (Table 1). We observed 604 generations encompassing 20 160 years. These values give an average 33.38 years generation duration time (95% CI: 32.76, 34.00).
Diversity indices, mismatch distribution, Fst
In general, standard within-population diversity indices show that PAR and SGP share similar levels of internal genetic variation (Supplementary Table S4). The only exception is Y-SNP-based Gene Diversity, which is significantly higher in PAR. Analogously, mismatch distributions (mtDNA) are almost identical in both samples, suggesting that these populations have experienced the same recent demographic changes (Supplementary Figure S3). Such changes can be interpreted as a demographic increase of the population, which is perfectly in line with what is known for the investigated area for the last two centuries.
As for Y-chromosomal hgs (Table 2), R-U152 is the most represented lineage in both populations, albeit with a significantly (Fisher test: P=0.0363) much higher frequency in SGP (44.26 vs 27.27%). From the other side, PAR differentiates itself from SGP primarily because of the high frequency of hg I1-L22 (15.91%), which in turn is completely absent in SGP (Fisher test: P=0.0004). Hg J2-M67*, also, is significantly (Fisher test: P=0.0443) more frequent in PAR than in SGP (13.64 vs 3.28%). Accordingly, we observe a slight but significant differentiation between PAR and SGP (Fst=0.030; P<0.01).
Contrarily to Y-chromosome, mtDNA variation shows that PAR and SGP are not significantly differentiated from each other (Fst=−0.002, P-value>0.05). Because PAR appears to be different from SGP only from the paternal point of view, all the following analyses are meant to explore such Y-chromosomal differentiation.
MDS
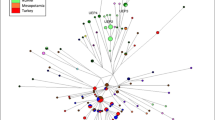
To check the position of PAR and SGP within a European Y-chromosomal genetic landscape, we performed an MDS analysis based on Y-STR haplotypes (Figure 1). Results show clearly that both populations, despite being significantly different from each other, fall within the variability spectrum of Italian populations.

Non-metric MDS representation of PAR, SGP and reference populations based on Y-STR data.
DAPC and dating
Y-chromosomal hg I1-L22 is the distinctive mark of PAR, being highly frequent here (15.91%) whereas completely absent in SGP. A DAPC-based exploration of I1-L22 haplotypes from PAR and from comparison European populations revealed three well-differentiated clusters (Figure 2, Supplementary Table S5). Interestingly, all PAR I1-L22 haplotypes fall within the same cluster (cluster 2) together with a few haplotypes from South-Eastern Italy (1), Germany (1), Poland and Slovakia (4). Other clusters (1 and 3) are composed mainly by haplotypes from Germany and Poland, but cluster 1 includes most of the I1-L22 haplotypes from South-Eastern Italy and the Balkans, while cluster 3 is enriched in Brabant and North-Western Italy haplotypes. These results suggest that I1-L22 individuals from PAR share a recent common ancestor that could have been living around the age when the Partecipanza was established. Accordingly, we estimated: (i) the age of I1-L22 PAR haplotypes and (ii) the age of whole cluster 2. The first estimate can be interpreted as a lower limit to the age of foundation of Partecipanza, while the second as an upper limit. For the calculations we use our own estimates of generation duration times and average STR mutation rate, as well as locus-specific Ballantyne et al. (2010) rates. Our results (Table 3) place the lower limit at around 600–700 years ago, while the upper limit is at around 1300–1400 years ago.

DAPC of Y-STR variation in I1-L22 haplotypes from PAR and reference populations. Scatterplot of the first and the second discriminant functions. Symbol size is proportional to the frequency of Y-STR haplotypes.
As for hg J2-M67*, comparing PAR and SGP haplotypes with those from European reference populations with DAPC, we obtain three different clusters (Supplementary Figure S4). One of them (cluster 3) is exclusively found in PAR, where it is associated to a single documented pedigree (P05, Table 1) including eight individuals. Another one (cluster 2) is found both in PAR (four haplotypes) and SGP (one haplotype), as well as in Germany and Southern Italy. Dates for the whole of cluster 2 (Table 3) are as much old as 6000–9000 years ago. Cluster 1, despite being well represented both in Germany and in Italy, has only one haplotype in SGP.
A DAPC-based comparison of PAR and SGP R-U152 haplotypes with those from reference European populations revealed five different clusters (Supplementary Table S5, Supplementary Figure S4). Each of these clusters is represented both in PAR and SGP. By pooling together clusters 1 and 2—which largely overlapped in our DAPC representations—we were able to date R-U152 haplotypes both in PAR and SGP. Results were quite similar (Table 3), pointing at a period between 3500 and 4500 years ago in both cases. Analogously, when dating cluster 3 considering both haplotypes from PAR and SGP (Table 3), we obtained around 3000–5000 years from present.
Discussion
In this study, we show that it is possible to shed light on important events of the recent genetic history of a region by carefully selecting the investigated population and the individuals to be sampled. Our PAR and SGP samples not only help to understand poorly known aspects of the genetic history of Italy and Europe, but they also offer important glimpses on issues of more general interest, such as the estimation of essential parameters such as Y-STR mutation rates and average generation duration time.
First of all, we observed that the peculiar rules that determine the affiliation to Partecipanza tend by themselves to minimize the possible discrepancies between Y-chromosomes, surnames and paternal pedigrees (King and Jobling, 2009b). For example, adoptive sons and the children of unmarried mothers would still bear a Partecipanza surname, but not the corresponding Y-chromosome lineage. In both cases, these individuals and their offspring would lose the right to participate. External Y-chromosomal lineages may introgress Partecipanza only through hidden adoptions and non-paternity events. However, the historical rate estimated for these events is equal to 1.12% per generation. This value is close to the one calculated with the same method by Larmuseau et al. (2014) for Flanders (0.91%) and it is substantially lower than the 8–30% per generation reported in behavioural studies (Larmuseau et al., 2014 and bibliography therein). These values are low enough to conclude that they did not substantially affect the Y-chromosomal genetic composition of the Partecipanza over the last centuries.
Our first aim was to test whether the idiosyncratic social-economic structure of Partecipanza could influence its own genetic variability. Cultural features are one of the most discussed determinants of genetic variation in human populations. The case of Partecipanza shows that being part of an economically advantaged élite (i.e., to be a Partecipante) may actually generate significant genetic differences with the rest of the population. It is important to consider that such peculiarities are limited to Y-chromosome variation and are not found in mtDNA. This result is in line with the specific transmission mechanism of the status of Partecipante that is strictly and exclusively male-mediated. This observation, together with the lack of any significant variability reduction in PAR compared with SGP (both for Y-chromosome and mtDNA), suggests that Partecipanza did not experience significant drift effects and the Y-chromosomal composition of PAR cannot be explained exclusively by isolation. Furthermore, our MDS analysis (Figure 1) clearly showed that both PAR and SGP lie within the space defined by Italian Y-chromosomal genetic variability.
These facts lead us to our second aim that was to reconstruct the time and the genetic origin of Partecipanza, as well as its implications for the genetic history of Italy and Europe. Differences between PAR and SGP are mainly determined by three Y-hgs: I1-L22, J2-M67* and R-U152. Frequency of Y-hg I1-L22 reaches 15.91% in PAR, being completely absent in SGP and rare in Italy (0.79%; Boattini et al., 2013). Furthermore, its frequency does not exceed 5% in all the considered comparison populations. As in the case of its parental clade I1-M253, I1-L22 is most frequently found in Northern Europe, around the Baltic sea, where it probably originated (Soares et al., 2010; Underhill et al., 2007).
As shown by DAPC (Figure 2), I1-L22 haplotypes from PAR form a tight and coherent cluster, suggesting that they may share a common ancestor who lived around the period in which Partecipanza was founded. Further proof of this is the fact that such hg is at present found in four different surnames and in nine paternal pedigrees (Supplementary Table S2). Considering that, to the best of our present knowledge, Partecipanza is at least two or three centuries older than its own surnames, the fact that different surnames show similar Y-chromosomal haplotypes is most easily explained as a consequence of their genetic common ancestor. Another convergent line of reasoning is provided by our time estimates. Despite the fact that Y-STR-based estimates are a controversial issue, in this study, we were able to overcome at least some of the most important criticisms brought to such methods. First, our results strongly suggest that PAR I1-L22 haplotypes are not the result of historical (or geographical) stratification; on the contrary, they are tightly related and seem to mark a single historic event. As a second, important point, we were able to estimate reliable values for parameters such as STR mutation rates and generation duration time directly from our data. Despite relatively wide CIs (Table 3), our estimates confirm with what is currently known about the historic origins of Partecipanza, pointing at a period comprised between 1300 and 600 years ago. Interestingly, the upper bound coincides with the Migration Period, and in particular with the settling of Ostrogoths and Lombards in Italy (493 AD and 568 AD, respectively). The lower bound might instead correspond to the devastating ‘Black Death’ epidemic of the fourteenth century (∼1350 AD), a strong bottleneck that may have affected our population. It is worth noting that the geographic distribution of hg I1-L22 and its alleged place of origin are consistent with the alleged route followed by some German peoples—Lombards and Goths in particular—from the Baltic shores to Italy.
The area of San Giovanni in Persiceto became part of the Lombard kingdom relatively late (728 AD, under King Aistulf) and their rule lasted only half a century, after having been defeated by the French King Charlemagne in 776 AD (Capitani, 2009). Nevertheless, a number of historical facts seem to link this area with Lombard settlements. Among them, the most relevant involves the Abbey of Nonantola, one of the most powerful monastic centres of the area. The Abbey was founded by Lombard kings in 752 AD (Bottazzi, 2003). Since then, emphyteutic grants from Nonantola (first documented in 1170 AD) had a relevant role in the formation of the assets of Partecipanza. San Giovanni in Persiceto, according to some scholars, was the seat of a Lombard Duchy in the second half of the eighth century (Bottazzi, 2003; Santos Salazar, 2006). Eventually, the case of a burial site discovered in the early 1960s but originally misinterpreted as a recent mass grave has been reported. This funerary evidence exhibits a number of Germanic features and has been recently radiocarbon dated to ∼1000 years ago (D'Adamo and Pedrini, 2013).
Putting it all together, it seems plausible to relate historical and archaeological information with our molecular results, suggesting that a Lombard component may have had a key role in the foundation of Partecipanza.
As it is obvious, we are not implying that hg I1-L22 coincides with ancient Lombards. Their original genetic variability is still unknown and probably varied in time and space. It is anyway reasonable to believe that I1-L22 was an important part of this background. Therefore, Partecipanza is not a Lombard ‘living fossil’. On the contrary, hgs J2-M67* and R-U152 reveal different aspects of its formative history. Contrarily to I1-L22, DAPC showed that J2-M67* PAR haplotypes form two different clusters (Supplementary Table S5, Supplementary Figure S4), suggesting that their high frequency (compared with SGP) is the result of historic/geographic stratification. One of these clusters (cluster 2) is represented both in PAR and SGP and is frequent in Italy and Germany, yielding a date as old as 6000 years before present (Table 3). Another one (cluster 3) is found only in PAR, coinciding with a single pedigree (P05: Supplementary Table S5, Supplementary Figure S4) whose common ancestor lived in the sixteenth century. A search through the YHRD database (www.yhrd.org) showed that its modal haplotype matches with one observed in Apulia (Southern Italy).
R-U152 is the most frequent hg both in PAR and SGP, as well as in the whole of North-Western Italy (Boattini et al., 2013). Although its origins are still unclear (Myres et al., 2011; Busby et al., 2012), its presence in the area of San Giovanni in Persiceto is much older than Partecipanza itself. Our dating experiments yielded dates ranging between 4000 and 5000 years ago for both populations (Table 3). In addition, R-U152 shows clear evidences of stratification both in PAR and SGP. In fact, DAPC revealed five different clusters, each of them including haplotypes from both populations (Supplementary Table S5, Supplementary Figure S4)
Our last goal was to estimate Y-STR mutation rates and average generation duration time. As far as the former is concerned, relevant literature provides contrasting estimates, which can be divided as follows:
-
1)
Evolutionary rate. First proposed by Zhivotovsky et al. (2004), such rate is inferred from hg variation in populations whose short-time history is known (foundation events) and is equal to 6.9 * 10−4 per STR per generation. It is widely used for dating purposes.
-
2)
Germline rates. Such rates are based on direct counts of mutational events in father-son pairs. Estimates in Ballantyne et al. (2010) are based on ∼2000 pairs and, for the 17 Y-STR set here considered, they average to 3.2 * 10−3 mutations per STR per generation.
-
3)
Pedigree rates. These rates are based on counts of mutational events within pedigrees. King and Jobling (2009a), by relying on a total of 274 generations (within 14 pedigrees) and 17 STRs, calculated a rate of 1.5 * 10−3.
Compared with King and Jobling (2009a), our calculations are based on a wider set of data (368 generations within 15 pedigrees) and our pedigrees do not include tight relatives. Although the number of studied pedigrees and the range of observed Y-chromosomal hgs is too limited to make strong conclusions, we observed that our estimate (3.90 * 10−3; 95% CI: 2.44 * 10−3, 5.68 * 10−3) is higher than King and Jobling's (2009a) and closer to Ballantyne et al. (2010) germline rate (which in fact is comprised in our CI). In addition, all these rates are an order of magnitude higher than Zhivotovsky et al. (2004) evolutionary rate. Following another line of evidence, Balanovsky et al. (2011) observed that time estimates based on germline rates have a good fit with dates obtained from linguistic and archaeological evidence, while dates based on evolutionary rates tend to be older. These facts suggest that Y-STR diversity is generally distributed in pedigrees following the germline rate, while evolutionary rate is much slower.
Turning now to the average duration of generation time, our estimate (33.38 years; 95% CI: 32.76, 34.00) is comparable to the 35-year value that King and Jobling (2009a) suggest as ideal for Britain. As a consequence, the 25-year value often used for time estimates and coalescent simulations does not seem to be a realistic estimate for paternal lineages in European populations.
Conclusion
In this study, we showed that a well-conceived sampling strategy (population choice, selection of individuals) facilitates the identification of otherwise hidden population events and historical stratifications. While doing this, we generated relevant information on issues that transcend the regional setting of this study.
The first aim of this study was to check if socio-economic stratification within the same population can determine specific genetic structures. Our results show that this is the case for our PAR and SGP samples. In particular, such structure is related to patrilines, whereas mtDNA showed no difference between the two samples. This agrees with the transmission of the shared assets within Partecipanza. Finally, by comparing PAR with SGP, as well as with Italian and European populations, we concluded that most of the Partecipanza founders bore Y-chromosome lineages that were already present in the area by a long time. Anyway, it seems plausible that such nucleus was in some way enriched by a Lombard component that settled here during the eighth century AD and continued to live there even after the defeat of the Lombard kingdom in 776 AD. Partecipanza, thanks to its particular set of rules, preserved (and possibly amplified) such a ‘trace’ that went lost in the open population living in the same area.
Our last result was the determination of important parameters such as Y-STR mutation rates and average generation duration time. Thanks to a combination of paternal pedigrees and Y-chromosome molecular data, we estimated a Y-STR mutation rate equal to 3.90 * 10−3 (17 STRs). Such rate is one order of magnitude higher than evolutionary rate (Zhivotovsky et al., 2004) and very similar to germline rates (Ballantyne et al., 2010). As for generation duration time, our estimate (33.38 years) is in line with what is known for comparable European populations (UK; King and Jobling, 2009a). These results have important consequences on currently debated issues, such as Y-chromosome molecular dating.
Future developments of our research will further explore some of the most important results of this study. The genetic structure of Partecipanza will be further tested with autosomal markers. As far as its ancestry is concerned, we will replicate our experiment by extending our sampling to other similar institutions. Turning instead to Y-chromosome mutation rates, comparisons between Y-haplotypes and genealogies will be extended to a higher number of Y-chromosome markers and paternal pedigrees.
Data archiving
Y-chromosome data (17 STRs and hgs) have been submitted to the Dryad Digital Repository: doi:10.5061/dryad.26qn0. HVS-I and HVS-II mtDNA sequences have been submitted to GenBank: accession numbers KM232757—KM232905.
Accession codes
References
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N . (1999). Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23: 147.
Balanovsky O, Dibirova K, Dybo A, Mudrak O, Frolova S, Pocheshkhova E et al. (2011). Parallel evolution of genes and languages in the Caucasus region. Mol Biol Evol 28: 2905–2920.
Ballantyne KN, Goedbloed M, Fang R, Schaap O, Lao O, Wollstein A et al. (2010). Mutability of Y-chromosomal microsatellites: rates, characteristics, molecular bases, and forensic implications. Am J Hum Genet 87: 341–353.
Behar DM, Van Oven M, Rosset S, Metspalu M, Loogväli EL, Silva NM et al. (2012). A "Copernican" reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet 90: 675–684.
Boattini A, Griso C, Pettener D . (2011b). Are ethnic minorities synonymous for genetic isolates? Comparing Walser and Romance populations in the Upper Lys Valley (Western Alps). J Anthropol Sci 89: 161–173.
Boattini A, Lisa A, Fiorani O, Zei G, Pettener D, Manni F . (2012). General method to unravel ancient population structures through surnames, final validation on Italian data. Hum Biol 84: 235–270.
Boattini A, Luiselli D, Sazzini M, Useli A, Tagarelli G, Pettener D . (2011a). Linking Italy and the Balkans. A Y-chromosome perspective from the Arbereshe of Calabria. Ann Hum Biol 38: 59–68.
Boattini A, Martinez-Cruz B, Sarno S, Harmant C, Useli A, Sanz P et al. (2013). Uniparental markers in Italy reveal a sex-biased genetic structure and different historical strata. PLoS ONE 8: e65441.
Bottazzi G . (2003). Monteveglio e Nonantola tra bizantini e longobardi. In: Cerami D, (ed) Monteveglio e Nonantola: Abbazie e Insediamenti Lungo le vie Appenniniche. Centro Studi Storici Nonantolani: Nonantola. pp 33–66.
Busby GB, Brisighelli F, Sánchez-Diz P, Ramos-Luis E, Martinez-Cadenas C, Thomas MG et al. (2012). The peopling of Europe and the cautionary tale of Y chromosome lineage R-M269. Proc Biol Sci 279: 884–892.
Capitani O . (2009) Storia dell'Italia Medievale. Laterza: Roma-Bari.
Capocasa M, Anagnostou P, Bachis V, Battaggia C, Bertoncini S, Biondi G et al. (2014). Linguistic, geographic and genetic isolation: a collaborative study of Italian populations. JASs 92: 201–231.
D'Adamo C, Pedrini W . (2013) I 34 scheletri del Poggio. Cronaca di una scoperta archeologica, Maglio Editore: S. Giovanni in Persiceto.
Excoffier L, Laval G, Schneider S . (2007). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Ferri G, Alù M . (2012) Development of six Y-SNPs assay for forensic analysis in European population. DNA in Forensics 2012, 5th International EMPOP Meeting-8th International Forensic. Y-User Workshop, Innsbruck.
Jobling MA . (2012). The impact of recent events on human genetic diversity. Phil Trans R Soc B 367: 793–799.
Jombart T, Devillard S, Balloux F . (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics 11: 94.
Jombart T . (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405.
Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF . (2008). New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 18: 830–838.
King TE, Jobling MA . (2009a). Founders, drift, and infidelity: the relationship between y chromosome diversity and patrilineal surnames. Mol Biol Evol 26: 1093–1102.
King TE, Jobling MA . (2009b). What's in a name? Y-chromosomes, surnames and the genetic genealogy revolution. Trends Genet 25: 351–360.
Komsta L . (2011) outliers: Tests for outliers. R package version 0.14 http://CRAN.R-project.org/package=outliers.
Lacan M, Keyser C, Crubézy E, Ludes B . (2012). Ancestry of modern Europeans: contributions of ancient DNA. Cell Mol Life Sci 70: 2473–2487.
Larmuseau MH, Vanderheyden N, Jacobs M, Coomans M, Larno L, Decorte R . (2011). Micro-geographic distribution of Y-chromosomal variation in the central-western European region Brabant. Forensic Sci Int Genet 5: 95–99.
Larmuseau MHD, Van Geystelen A, van Oven M, Decorte R . (2013). Genetic Genealogy Comes of Age: Perspectives on the Use of Deep-Rooted Pedigrees in Human Population Genetics. Am J Phys Anthropol 150: 505–511.
Larmuseau MHD, Vanoverbeke J, Van Geystelen A, Defraene G, Vanderheyden N, Matthys K et al. (2014). Low historical rates of cuckholdry in a Western European human population traced by Y-chromosome and genealogical data. Proc Biol Sci 280: 20132400.
Laval G, SanCristobal M, Chevalet C . (2002). Measuring genetic distances between breeds: use of some distances in various short term evolution models. Genet Sel Evol 34: 481–507.
Martínez-Cruz B, Harmant C, Platt DE, Haak W, Manry J, Ramos-Luis E et al. (2012). Evidence of pre-Roman tribal genetic structure in Basques from uniparentally inherited markers. Mol Biol Evol 29: 2211–2222.
Myres NM, Rootsi S, Lin AA, Järve M, King RJ, Kutuev I et al. (2011). A major Y-chromosome haplogroup R1b Holocene era founder effect in Central and Western Europe. Eur J Hum Genet 19: 95–101.
Onofri V, Alessandrini F, Turchi C, Pesaresi M, Buscemi L, Tagliabracci A . (2006). Development of multiplex PCRs for evolutionary and forensic applications of 37 human Y chromosome SNPs. Forensic Sci Int 57: 23–35.
R Core Team. (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria http://www.R-project.org/.
Rębała K, Martínez-Cruz B, Tönjes A, Kovacs P, Stumvoll M, Lindner I et al. (2013). Contemporary paternal genetic landscape of Polish and German populations: from early medieval Slavic expansion to post-World War II resettlements. Eur J Hum Genet 21: 415–422.
Regueiro M, Rivera L, Damnjanovic T, Lukovic L, Milasin J, Herrera RJ . (2012). High levels of Paleolithic Y-chromosome lineages characterize Serbia. Gene 498: 59–67.
Reynolds JB, Weir BS, Cockerham CC . (1983). Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics 105: 767–779.
Santos Salazar I . (2006). Castrum Persiceta: Potere e territorio in uno spazio di frontiera dal secolo VI al IX. Reti Medievali Rivista 7: 1–20.
Sarno S, Boattini A, Carta M, Ferri G, Alù M, Yang Yao D et al. (2014). An ancient Mediterranean melting pot: investigating the uniparental genetic structure and population history of Sicily and Southern Italy. PLoS ONE 9: e96074.
Sengupta S, Zhivotovsky LA, King R, Mehdi SQ, Edmonds CA, Chow CE et al. (2006). Polarity and temporality of high-resolution Y-chromosome distributions in India identify both indigenous and exogenous expansions and reveal minor genetic influence of Central Asian Pastoralists. Am J Hum Genet 78: 202–221.
Soares P, Achilli A, Semino O, Davies W, Macaulay V, Bandelt HJ et al. (2010). The Archaeogenetics of Europe. Curr Biol 20: R174–R183.
Underhill PA, Myres NM, Rootsi S, Chow CT, Lin AA, Otillar RP et al. (2007). New phylogenetic relationships for y-chromosome haplogroup i: reappraising its phylogeography and prehistory. In: Mellars P, Boyle K, Bar-Yosef O, Stringer C, (eds) Rethinking the Human Revolution. McDonald Institute for Archaeological Research: Cambridge (UK). pp 33–42.
van Oven M, Van Geystelen A, Kayser M, Decorte R, Larmuseau MH . (2014). Seeing the wood for the trees: a minimal reference phylogeny for the human Y chromosome. Hum Mutat 35: 187–191.
Zanarelli M . (1992). I beni comuni e le forme di gestione attuate dalle comunità rurali: il caso di San Giovanni in Persiceto e di Medicina. In: Fregni, (ed) Terre e Comunità nell'Italia Padana. Il caso delle Partecipanze Agrarie Emiliane: da beni comuni a beni collettivi. Edizioni Centro Federico Odirici: Mantova. pp 147–173.
Zhivotovsky LA, Underhill PA, Cinnioglu C, Kayser M, Morar B, Kivisild T et al. (2004). The effective mutation rate at Y chromosome short tandem repeats, with application to human population-divergence time. Am J Hum Genet 74: 50–61.
Acknowledgements
This research was made possible by grants from the Italian Ministry of Education, University and Research (PRIN projects 2007 and 2009, Principal Investigator DP). SS was supported by the European Research Council ERC-2011-AdG 295733 grant (Langelin). We thank all the volunteers who kindly accepted to participate to this study. We are indebted with the Personnel of the ‘Consorzio dei Partecipanti di San Giovanni in Persiceto’, the Municipality of San Giovanni in Persiceto and the local AVIS (Associazione Volontari Italiani del Sangue) centre for helping us to design the sampling campaign and to collect the archival data. Special thanks are due to Vittorio Cocchi (President of the ‘Consorzio dei Partecipanti’), Raffaella Scagliarini (Responsible of the Historical Archive of the ‘Consorzio dei Partecipanti’), Renato Mazzuca (Mayor of San Giovanni in Persiceto) and Sonia Camprini (Council Member of San Giovanni in Persiceto). Last but not least, we thank Dr Eugenio Bortolini for his valuable comments and suggestions to improve the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Boattini, A., Sarno, S., Pedrini, P. et al. Traces of medieval migrations in a socially stratified population from Northern Italy. Evidence from uniparental markers and deep-rooted pedigrees. Heredity 114, 155–162 (2015). https://doi.org/10.1038/hdy.2014.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.77
This article is cited by
-
Genetic history of Calabrian Greeks reveals ancient events and long term isolation in the Aspromonte area of Southern Italy
Scientific Reports (2021)
-
Estimating Y-Str Mutation Rates and Tmrca Through Deep-Rooting Italian Pedigrees
Scientific Reports (2019)
-
The Y chromosome as the most popular marker in genetic genealogy benefits interdisciplinary research
Human Genetics (2017)
-
The Italian genome reflects the history of Europe and the Mediterranean basin
European Journal of Human Genetics (2016)
-
Y-chromosome diversity in Catalan surname samples: insights into surname origin and frequency
European Journal of Human Genetics (2015)

{kind=link}
{kind=link}
{kind=link}