
Abstract
Purpose:
Routine screening for evidence of DNA mismatch repair abnormalities can identify colorectal cancer patients with Lynch syndrome, but impact in usual care settings requires study. After implementing routine screening at our university and safety-net health systems as usual practice, our aims were to determine outcomes, including screening process quality.
Methods:
We conducted a retrospective cohort study from 1 May 2010 to 1 May 2011. Screening included reflexive immunohistochemistry to evaluate DNA mismatch repair protein expression for patients with colorectal cancer aged ≤70 years, with a cancer genetics team following up results. Screening outcomes, as well as challenges to a high-quality screening process were evaluated.
Results:
We included 129 patients (mean age 56 years, 36% female); 100 had immunohistochemistry screening completed. Twelve patients had abnormal immunohistochemistry: four with definite Lynch syndrome, four with probable Lynch syndrome, and three without Lynch syndrome; one patient had an incomplete work-up. Lynch syndrome was confirmed for 6/13 asymptomatic relatives tested. Screening process quality was optimal for 77.5% of patients. Barriers to optimal quality screening included ensuring reflexive immunohistochemistry completion, complete follow-up of abnormal immunohistochemistry, and timely incorporation of results into clinical decision making.
Conclusion:
Usual care implementation of routine screening for Lynch syndrome can result in significant rates of detection, even in a largely safety-net setting. To optimize implementation, challenges to high-quality Lynch syndrome screening, such as ensuring reflexive screening completion and clinically indicated genetic testing and follow-up for abnormal screens, must be identified and addressed.
Genet Med 15 12, 925–932.
Similar content being viewed by others
Main
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and third leading cause of cancer death in the United States.1 Three to five percent of all newly diagnosed CRCs are attributable to Lynch syndrome (LS) (also known as hereditary nonpolyposis colorectal cancer).2,3 LS is caused by a germline mutation in one of the four DNA mismatch repair proteins (MLH1, MSH2, PMS2, or MSH6) that results in progressive accumulation of genetic defects conferring cancer susceptibility.4 Mutations are autosomal dominant and result in high lifetime risk for CRC, endometrial cancer, and other cancers.4,5 Identification of CRC patients with LS may optimize CRC treatment and cancer surveillance,6,7,8,9 and allow for early identification of family members who are asymptomatic carriers that results in reduced cancer-associated morbidity and mortality.10,11,12
Recently, there has been marked interest in screening all patients with newly diagnosed CRC for LS through routine laboratory testing.3,4,13,14,15 In this approach, a patient’s CRC specimen is screened for pathologic or molecular features consistent with LS, and, if the screen is abnormal, the patient is referred for genetic counseling and confirmatory testing. Screening for evidence of mismatch repair can be completed by testing for presence of high microsatellite instability or by testing for abnormal absent expression of one or more of the four DNA mismatch repair proteins affected by LS using immunohistochemistry (IHC).3,16
Prior research has demonstrated that the universal screening approach is 69–100% sensitive and 80–94% specific for identifying patients with LS 3,4,17 Moreover, this approach may identify patients who would not be identified as having high risk for LS on the basis of clinical criteria such as the Bethesda guidelines, which require a clinician to screen for LS by evaluating age at presentation, family history, and morphologic features of a patient’s CRC specimen.3,4,18,19
The Centers for Disease Control and Prevention recently asked the Evaluation of Genomic Applications in Practice and Prevention Working Group, a panel of independent experts, to review evidence to support widespread universal screening of patients with CRC as an approach to identifying individuals with LS. The Evaluation of Genomic Applications in Practice and Prevention Working Group concluded that there was sufficient evidence to recommend offering genetic testing for LS to individuals with CRC, mainly because such testing may allow for detection of asymptomatic relatives who are LS carriers.15 Subsequently, an international panel of LS experts has supported the universal screening strategy,13 evidence supporting effectiveness of universal testing for identifying CRC patients with LS has been strengthened,3 and a recent analysis concluded that a universal screening approach employing IHC for absence of one of the four DNA mismatch repair proteins is cost effective.12,20,21
Despite enthusiasm for the universal screening approach, outside of specialized cancer centers, few groups have implemented screening programs,22 and little is known about how to ensure high-quality screening processes.14 Universal screening is challenging because multiple steps are involved in the screening process.15 CRC resections or biopsies must be systematically subjected to microsatellite instability and/or IHC testing. Abnormal screening results must be followed up with genetic counseling, and often genetic testing. Genetic work-up results need correct interpretation and may need to be incorporated into clinical care in timely fashion, such as to allow planning for subtotal versus extended colectomy if indicated.7,8 Given the complexity of universal screening, and a lack of reports on implementing this approach, more data are required to understand the ultimate potential impact of widespread implementation. Experience with other complex screening processes such as breast cancer screening has demonstrated that the ultimate effectiveness of intended screening may be highly dependent on how well cancer screening processes are implemented.14,23,24
In 2010, we implemented a routine screening protocol at our university and safety-net health systems with the goal of screening all patients with CRC aged 70 and younger for absence of one of the four DNA mismatch repair proteins associated with LS using IHC, and completing all clinically indicated follow-up for patients with abnormal screening results. In this report, our aims were to determine the impact of implementing this protocol on the detection of patients with LS, and to describe the challenges to successful implementation of routine screening we encountered.
Materials and Methods
Study setting
We conducted a retrospective cohort study of all patients diagnosed with CRC since the initiation of routine screening at UT Southwestern Medical Center, as well as at Parkland Health and Hospital System, both in Dallas, Texas. UT Southwestern is a 270-bed tertiary care university health system that mainly cares for insured patients and is home to the National Cancer Institute–designated Simmons Cancer Center. Parkland is a 960-bed tertiary and safety-net health system that is the main provider of health care for uninsured and Medicaid patients residing in Dallas County, TX. Parkland is also closely affiliated with UT Southwestern’s Cancer Center and shares many primary- and specialty-care providers. Both UT Southwestern and Parkland maintain cancer registries that have been approved by the American College of Surgeons Commission on Cancer.
Routine testing protocol
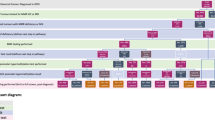
We initiated our screening protocol ( Figure 1 ) for LS at both Parkland and UT Southwestern beginning 1 May 2010. The protocol recommended testing all CRC biopsies and resection specimens from patients aged ≤70 years for presence of one of the four DNA mismatch repair proteins (MLH1, MSH2, MSH6, and PMS2), consistent with recommendations from an international consensus conference.13 The intent of our protocol was to have IHC reflexively ordered by the pathologist after any CRC diagnosis. IHC was performed as previously described, using automated image analysis, with staining of <30% of cells considered abnormal.25 Antibodies used for IHC were MLH1 (clone, G168-728, 1:300, BD Biosciences, San Jose, CA), MSH2 (clone, G219-1129, 1:20, Cell Marque, Rocklin, CA), MSH6 (clone, 44, 1:100, Cell Marque), and PMS2 (clone, A16-4, dilution 1:250, BD Biosciences).

Dallas routine screening protocol for Lynch syndrome. Our protocol specified reflex IHC screening for all patients with CRC aged ≤70 years, with reporting of IHC results to the medical record and genetic counselors on a weekly basis. Genetic counselors served as the “quarterback” for all testing, ensuring contact and follow-up of all patients with abnormal results. After initiating the protocol, a suboptimal rate of reflexive IHC was noted, and an added step, in which the genetics team monitored all new CRC diagnoses on a weekly basis to ensure IHC completion was added (see text). CRC, colorectal cancer; IHC, immunohistochemistry; NCCN, National Comprehensive Cancer Network.
No further follow-up was required if expression of all four DNA mismatch repair proteins was normal. If IHC results were abnormal, the patient was contacted by a genetic counselor to schedule a counseling visit to discuss further testing. The testing laboratory submitted a weekly report of all IHC results to the genetic counselors, who contacted patients with abnormal results to schedule counseling visits. At the genetic counseling visit, a complete family history was taken, and further genetic testing was pursued as recommended by National Comprehensive Cancer Network clinical practice guidelines for management of patients with suspected LS.6 Analysis of the mismatch repair genes performed at Myriad Genetic Laboratories (Salt Lake City, UT) and Mayo Medical Laboratories (Rochester, MN) consisted of full-sequence determination by long-range and nested PCR of all genes performed in both forward and reverse directions comprising all exons and adjacent noncoding intronic base pairs for the MLH1, MSH2, MSH6, and PMS2 genes. Myriad Genetic Laboratories analysis of deletions and duplications consisted of microarray comparative genomic hybridization analysis of all coding exons of MLH1, MSH2, MSH6 and the clinically relevant 3′ region of the EPCAM gene. Deletions and duplications of the PMS2 gene were assessed using multiplex ligation-dependent probe amplification at Myriad Genetic Laboratories. Mayo Medical Laboratories analysis of deletions and duplications consisted of gene dosage analysis by either multiplex ligation-dependent probe amplification or Southern blot of all MMR genes and EPCAM. BRAF V600E and MLH1 promoter methylation analyses were performed at ARUP Laboratories (Salt Lake City, UT). The BRAF V600E mutation analysis consisted of DNA isolation from microdissected tumor tissue, amplification for exon 15, and use of pyrosequencing for mutation detection. MLH1 promoter methylation analysis consisted of DNA isolation from microdissected tumor tissue followed by amplification of a segment of the MLH1 promoter region using methylation-specific real-time PCR. The MLH1 methylation level is calculated by comparison to the amplification of a reference gene.
After discovering a high rate of missed reflexive IHC initiation after CRC diagnoses, we changed our protocol beginning February 2011 such that the genetics team monitored tumor board logs of all newly diagnosed cases of CRC and compared the list of tumor samples analyzed against the institution’s tumor registry. Our genetics team then alerted the pathology laboratory if IHC had not been completed and ensured that IHC was performed. Our routine screening protocol is summarized in Figure 1 .
Patient selection
For this report, we identified all patients aged 18–70 diagnosed with CRC at either health system from 1 May 2010 through 1 May 2011. Patients with CRC were identified by querying cancer registry records at both institutions. Patients who had CRC diagnosed at outside institutions, and no subsequent CRC biopsies or surgical resections at either health system, were excluded from analysis.
Data collection
During the study time period, both institutions used the comprehensive EPIC electronic medical record to record all patient-care activities, including operative reports, clinical progress notes, and laboratory results. For each patient, data abstracted from the electronic medical and cancer registry records included sex, race, ethnicity, age, date of initial diagnosis, site/location of tumor, Surveillance Epidemiology and End Results general summary stage, treatment timing, and vital status.
Outcome assessment
To examine the impact of implementation of universal screening for LS, we determined the outcomes of screening among patients with abnormal IHC testing and the rate of high-quality screening, and characterized challenges to high-quality screening.
Outcomes among patients with abnormal IHC screening. To determine the outcomes of routine screening among patients with abnormal IHC tests, we placed each patient into one of four mutually exclusive categories based on clinical work-up: (i) definite LS, (ii) no LS, (iii) probable LS, and (iv) incomplete work-up (see Table 1 for definitions). In addition, we recorded the number of known relatives of probands diagnosed with LS who underwent genetic testing and counseling, as well as the number of known relatives ultimately diagnosed with LS.
Rate of high-quality screening. High-quality LS screening was defined by one of two processes: (i) completing IHC screening with a normal result, or (ii) completing IHC screening with an abnormal result followed up with completion of genetic counseling and clinically indicated genetic testing. Suboptimal quality screening was defined by either (i) failure to complete IHC screening or (ii) failure to complete genetic counseling and clinically indicated genetic testing after having an abnormal IHC screening.
Challenges to high-quality screening. To characterize challenges to high-quality screening, we reviewed medical records—including clinical care notes—and attempted to determine reasons for suboptimal screening. In addition, we reviewed medical records to determine whether abnormal IHC results were recognized by the patient’s treating physicians and surgeons and incorporated into clinical care decisions. Finally, we searched to identify any adverse effects (such as psychological distress) that occurred as a result of routine screening.
All outcomes were characterized with descriptive statistics using STATA version 9.1 (College Station, TX) and Microsoft Excel. The study was approved by the UT Southwestern and Parkland institutional review boards.
Results
Of 159 CRC patients identified, 129 (81%) met inclusion criteria for this study. Among included patients, 29 were cared for primarily at UT Southwestern and 100 were cared for primarily at our Parkland safety-net hospital. The sample was 36.4% female, and the mean age was 55.7 years. Clinical characteristics of included patients are summarized in Table 2 . Of the 129 patients meeting inclusion criteria, 100 (77.5%) had IHC screening completed.
Outcomes of routine screening
Twelve patients had abnormal IHC screening ( Table 3 ); of these, four patients had definite LS ( Table 3 ). Of the four patients with LS, two were younger than 40 years of age at CRC diagnosis, two met Amsterdam criteria, and one met Bethesda guidelines, yet none had clinical suspicion for LS raised by treating physicians before completion of IHC screening.
Three patients with abnormal IHC screening clearly did not have LS, including one who had previously been diagnosed with Li Fraumeni syndrome ( Table 3 ).
Four patients had probable LS ( Table 3 ). Three of the four patients with probable LS had loss of MSH2/MSH6 expression on IHC with an uninformative work-up. The fourth patient with probable LS had a loss of expression of MLH1/PMS2 with normal MLH1 and PMS2 sequencing and negative BRAF mutation and MLH1 promoter hypermethylation analyses.
One patient with abnormal MSH2/MSH6 expression had an incomplete work-up ( Table 3 ). Therefore, we could not confirm whether or not the patient would fall into the definite or probable LS category.
Ten of 12 patients with abnormal IHC and all four patients with definite LS were diagnosed at our safety-net hospital. To date, 13 family members of the four probands with definite LS have undergone genetic testing and counseling (seven, two, and four relatives for probands A, B, and D, respectively). Of the 13 relatives tested, six were diagnosed with LS (two, one, and three relatives for probands A, B, and D, respectively).
Screening process quality and challenges
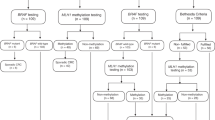
Among all patients with CRC (n = 129) who could have been candidates for screening, the screening process was optimal for 77.5% of patients. Figure 2 characterizes screening process success and failures for all patients. The most common failure was due to nonreflexive IHC testing by the pathology laboratory when we first implemented our protocol (n = 28); 23 of 28 failures occurred before having genetic counselors routinely confirm IHC testing for all new IHC diagnoses.

Screening process outcomes of routine screening for Lynch syndrome. Of 129 patients with CRC eligible for screening, 100 completed IHC screening. Reasons for screening noncompletion included failure to initiate reflex IHC (n = 28) and insufficient tissue for analysis (n = 1). Twelve patients had abnormal IHC, with 11 completing all clinically indicated follow-up. One patient with abnormal IHC was lost to follow-up despite multiple attempts to reach the patient by mail and phone. CRC, colorectal cancer; IHC, immunohistochemistry.
We noted several additional challenges to optimal delivery of routine screening. Five of 12 patients with abnormal IHC (cases B, C, E, J, and L) had sufficient colonoscopy biopsy tissue to perform screening before surgery but did not have testing performed until after surgery, on the resection specimen. Two of these patients (cases B and C) were ultimately diagnosed with LS but had undergone hemicolectomy by the time of diagnosis. Four of 12 patients (cases A, D, G, and H) with abnormal IHC had screening completed on colonoscopy biopsy presurgery, but IHC results were not optimally incorporated into surgical decision making. Case G was inappropriately offered extended colectomy before genetic testing for LS; ultimately, she refused and was subsequently confirmed to not have a mutation. Cases A and H had abnormal IHCs that were not resolved before surgery; case A was confirmed to have LS. Case D had optimal management, in that LS was diagnosed presurgery, and extended colectomy was offered to the patient, although he chose a more limited resection.
One patient faced significant logistical challenges after LS diagnosis. This patient was an uninsured, non–Dallas County resident, and therefore was not eligible for our health system’s medical assistance program for the uninsured. Thus, despite receiving a LS diagnosis and specific surveillance recommendations, he was unable to obtain annual colonoscopy surveillance or follow-up care at our health systems.
Discussion
We studied outcomes of implementation of a routine screening approach as part of usual CRC care for identifying patients with LS, as well as challenges to a high-quality screening process. After just 1 year, among 100 CRC cases that had IHC performed, we found four patients with definite and four patients with probable LS. Through testing relatives of our four probands with definite LS, an additional six mutation carriers were identified. The 4% rate of definite LS among patients with CRC we observed is very similar to the 3% rate reported by the largest pooled data analysis of strategies for detecting LS.3
Evaluation of our screening process revealed several challenges to high-quality routine screening, including an initial high failure rate to initiate reflexive screening, failure to consistently incorporate abnormal screening results into clinical decision making, and inability to follow up with one patient with an abnormal screen. Several screening process successes were also notable. For example, all but 1 of 12 patients with abnormal screening results received appropriate genetic counseling and testing in follow-up.
A high rate of appropriate follow-up occurred although nearly all of the patients were underserved patients cared for at our safety-net health system. The underserved, uninsured, and minority patients disproportionately cared for at safety-net health systems are often the last to benefit from innovations in cancer diagnosis and treatment—our report shows that routine screening can be delivered to this group, and that underserved patients can complete appropriate genetic testing and bring in unaffected relatives for genetic screening. We speculate that our success among underserved populations may be attributable to our model for delivering genetic counseling services. Rather than asking patients to come to a university cancer center clinic location separate from the safety-net health system campus, our genetics team visits patients in the same physical clinical location as where medical oncology consultations are provided. This allows us to use the same staff and facilities that our patients are familiar with, and may increase chances of successful follow-up. Our success in an underserved setting suggests that if clinicians and health systems caring for underserved populations offer routine screening for LS it may yield similar results to those observed in research settings.
Although multiple studies have demonstrated that the yield of universal screening for identifying patients with LS is both substantial and superior to the use of clinical criteria,3,4,13,19 our study addresses an important gap in the literature regarding challenges to successful implementation of routine screening in usual practice. The need to understand and address challenges to implementation of routine screening in usual practice has been highlighted by two expert groups.14,15 A literature review conducted to support a national Centers for Disease Control and Prevention conference on screening implementation15 and our own review identified just one published study of usual care challenges to high-quality screening.26 South et al.26 reported on implementation of universal screening in usual practice at their university medical center. Unexpectedly, only 27% of patients with abnormal screening completed clinically indicated genetic counseling. Ultimately, 2 patients were diagnosed with LS after screening 270 primary CRC patients. Overall, this study raised concerns regarding the ability to accomplish a high rate of clinically indicated genetic counseling and testing after initial abnormal screening, and suggests a need for approaches to ensure high rates of appropriate follow-up.
In addition to the concerns highlighted by the South et al.26 study, the Centers for Disease Control and Prevention conference attendees identified several additional potential challenges to universal screening, including lack of provider knowledge of LS, need for identification of a clinician with primary responsibility for the screening process, need for strategies for evaluating unaffected relatives, lag time between initial CRC diagnosis and molecular screening results, and patient and provider compliance with follow-up after screening.15
Our protocol and initial reported experience addresses many of these issues. First, we established the genetic counseling team as the “quarterback” for our universal screening protocol to ensure screening completion and subsequent follow-up of abnormal tests. We chose this approach because the genetics team is best positioned to execute best practices and advise patients and clinicians, and because the genetics team has the most vested clinical interest in identifying patients with potential LS. Second, we confirm the Centers for Disease Control and Prevention group’s concerns regarding potential for lag time between CRC diagnosis and availability of IHC screening results. Third, we can verify that provider interpretation and management of abnormal IHC results may be a significant issue, given that we found that several patients with abnormal IHC did not have IHC results appropriately incorporated into surgical planning. For example, there was one patient with abnormal IHC who was offered total colectomy before confirmation of LS, exposing the patient to potential harm. Fourth, we identified one uninsured patient with LS who could not be offered access to cancer surveillance. In addition to these concerns, we also note that initial requests for the pathology laboratory to initiate “reflexive” testing may not in practice be reflexive, but this can be addressed by close monitoring of new CRC diagnoses within an institution. Ultimately, although the potential benefit of implementing routine screening is promising, more research and experience is required to ensure that we can implement high-quality universal screening processes in the general population.14
Several limitations may be considered in interpreting our results. Our universal screening approach was limited to patients with CRC ≤70 years of age based on an international consensus published before the start of our efforts.13 Since then, at least one study has suggested improved yield for LS detection when universal screening without an age cutoff is used.3 As such, in our population, we may have underestimated the true prevalence of patients with abnormal IHC and LS. Such bias would have been unlikely, however, to impact our evaluation of the screening process quality. One patient (case F) had abnormal IHC, but further work-up was not pursued at the discretion of the primary medical team because she had previously been diagnosed with Li Fraumeni syndrome. Thus, we cannot exclude concurrent LS, although it would be extremely unlikely for a patient to carry both syndromes. Evaluation of impact of identifying LS carriers on clinical decision making did not extend to subsequent decisions on cancer screening/surveillance because the study time frame did not allow for these assessments. In contrast to results reported by the largest pooled analysis of LS screening,3 all of our patients with definite LS met Amsterdam or Bethesda criteria. Although we cannot say with certainty that these individuals would have been referred for screening and diagnosed without routine IHC, we think this would have been unlikely given that review of clinical notes suggests that teams caring for these patients were not actively recording and acting on family history risk information. Our cohort size was modest; larger studies of routine screening implementation, particularly including underserved populations, are needed to verify our results. Strengths of our study include observations from two separate health systems, with outcomes recorded for screening in usual practice, including careful evaluation of the quality of the screening process.
In summary, we successfully implemented a protocol for routine screening of patients with CRC for LS in university and safety-net health system settings. As a result, we identified four patients with definite, and four patients with probable LS within 1 year of protocol implementation, as well as six asymptomatic relatives who are mutation carriers. We also demonstrated a high rate of appropriate follow-up among patients with abnormal screening in a setting where most patients were underserved and cared for at a safety-net health system. Our screening process evaluation identified several challenges to high-quality implementation of universal testing, including ensuring completion of reflexive testing by the pathology laboratory, complete follow-up of all patients with abnormal screening results, and timely incorporation of screening results into clinical decision making. Despite these challenges, we found that an approach using the genetic team as the “quarterback” for ensuring reflexive testing completion and follow-up of abnormal screening can result in largely successful implementation of routine screening. Our findings can be used to aid in the development of usual care protocols for routine screening implementation and to inform future research in this area.
Disclosure
The authors declare no conflict of interest.
References
American Cancer Society. Cancer facts and figures. http://www.cancer.org/Research/CancerFactsFigures/index.
Lynch HT, de la Chapelle A . Hereditary colorectal cancer. N Engl J Med 2003;348:919–932.
Moreira L, Balaguer F, Lindor N, et al.; EPICOLON Consortium. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012;308:1555–1565.
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851–1860.
Bonadona V, Bonaïti B, Olschwang S, et al.; French Cancer Genetics Network. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305:2304–2310.
National Comprehensive Cancer Network. NCCN Guidelines. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp.
Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60:950–957.
Kalady MF, McGannon E, Vogel JD, Manilich E, Fazio VW, Church JM . Risk of colorectal adenoma and carcinoma after colectomy for colorectal cancer in patients meeting Amsterdam criteria. Ann Surg 2010;252:507–511; discussion 511–513.
Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 2006;354:261–269.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009;11:35–41.
Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000;118:829–834.
Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a cost-effectiveness analysis. Ann Intern Med 2011;155:69–79.
Boland CR, Shike M . Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology 2010;138:2197.e1–2197.e7.
Khoury MJ, Coates RJ, Fennell ML, et al. Multilevel research and the challenges of implementing genomic medicine. J Natl Cancer Inst Monographs 2012;2012:112–120.
Bellcross CA, Bedrosian SR, Daniels E, et al. Implementing screening for Lynch syndrome among patients with newly diagnosed colorectal cancer: summary of a public health/clinical collaborative meeting. Genet Med 2012;14:152–162.
Southey MC, Jenkins MA, Mead L, et al. Use of molecular tumor characteristics to prioritize mismatch repair gene testing in early-onset colorectal cancer. J Clin Oncol 2005;23:6524–6532.
Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN . EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 2009;11:42–65.
Singh H, Schiesser R, Anand G, Richardson PA, El-Serag HB . Underdiagnosis of Lynch syndrome involves more than family history criteria. Clin Gastroenterol Hepatol 2010;8:523–529.
van Lier MG, Leenen CH, Wagner A, et al.; LIMO Study Group. Yield of routine molecular analyses in colorectal cancer patients =70 years to detect underlying Lynch syndrome. J Pathol 2012;226:764–774.
Mvundura M, Grosse SD, Hampel H, Palomaki GE . The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med 2010;12:93–104.
Gudgeon JM, Williams JL, Burt RW, Samowitz WS, Snow GL, Williams MS . Lynch syndrome screening implementation: business analysis by a healthcare system. Am J Manag Care 2011;17:e288–e300.
Beamer LC, Grant ML, Espenschied CR, et al. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol 2012;30:1058–1063.
Taplin SH, Rodgers AB . Toward improving the quality of cancer care: addressing the interfaces of primary and oncology-related subspecialty care. J Natl Cancer Inst Monographs 2010;2010:3–10.
Zapka J, Taplin SH, Ganz P, Grunfeld E, Sterba K . Multilevel factors affecting quality: examples from the cancer care continuum. J Natl Cancer Inst Monographs 2012;2012:11–19.
Gupta S, Ashfaq R, Kapur P, et al. Microsatellite instability among individuals of Hispanic origin with colorectal cancer. Cancer 2010;116:4965–4972.
South CD, Yearsley M, Martin E, Arnold M, Frankel W, Hampel H . Immunohistochemistry staining for the mismatch repair proteins in the clinical care of patients with colorectal cancer. Genet Med 2009;11:812–817.
Acknowledgements
This work was supported by the Cancer Prevention and Research Institute of Texas (grant numbers PP100039 and PP120229 to S.G.) and the National Cancer Institute at the National Institutes of Health (grant number U54CA163308 to Celette Sugg Skinner (PI), S.G. (Co-I)). The authors thank Joan Cox, Alejandra Madrigales, and the entire staff of the UT Southwestern and Parkland Health and Hospital Cancer Registries for assistance with acquiring study data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Marquez, E., Geng, Z., Pass, S. et al. Implementation of routine screening for Lynch syndrome in university and safety-net health system settings: successes and challenges. Genet Med 15, 925–932 (2013). https://doi.org/10.1038/gim.2013.45
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.45
Keywords
This article is cited by
-
Comparison of universal screening in major lynch-associated tumors: a systematic review of literature
Familial Cancer (2022)
-
Universal tumor screening for Lynch syndrome: perspectives of Canadian pathologists and genetic counselors
Journal of Community Genetics (2019)
-
Successful implementation of Lynch syndrome screening in a safety net institution
Journal of Community Genetics (2016)
-
Screening for Lynch Syndrome: It Is Time to Shift the Focus
Digestive Diseases and Sciences (2015)
