
Abstract
Purpose:
The aim of this study was to examine Lynch syndrome screening of patients with metastatic colorectal cancer in integrated health-care-delivery organizations.
Methods:
We determined the availability of Lynch syndrome screening criteria and actual Lynch syndrome screening in the medical records of 1,188 patients diagnosed with metastatic colorectal cancer between 2004 and 2009 at seven institutions in the Cancer Research Network.
Results:
We found infrequent use of Lynch syndrome screening (41/1,188). Family history was available for 937 of the 1,188 patients (79%). There was sufficient information to assess Lynch syndrome risk using family history–based criteria in 719 of the 937 patients (77%) with family history documentation. In 391 individuals with a family history of a Lynch syndrome–associated cancer, 107 (27%) could not be evaluated due to missing information such as age of cancer onset. Eleven percent of patients who met the Bethesda criteria and 25% of individuals who met the Amsterdam II criteria were screened for Lynch syndrome. Recommended guidelines were adhered to during screening, but no testing method was preferred.
Conclusion:
The information required for Lynch syndrome screening decisions is routinely collected but seldom used. There is a critical gap between collection of family history and its use to guide Lynch syndrome screening, which may support a case for implementation of universal screening guidelines.
Genet Med 15 12, 933–940.
Similar content being viewed by others
Main
Lynch syndrome (LS), a hereditary cancer syndrome that accounts for 2–5% of the ~103,000 annual colorectal cancer (CRC) diagnoses in the United States,1,2,3,4,5,6 confers an increased risk of developing a number of other cancers including gastric, endometrial, ovarian, sebaceous gland, breast, and prostate cancers, transitional carcinoma of the ureter, and glioblastoma.3,4,6,7,8
LS is caused by autosomal dominant mutations in any one of four mismatch repair genes (MLH1, MLH2, MLH6, and PMS2).3,4 Individuals who harbor these germline mutations have an increased lifetime risk of developing CRC before age 50,9,10 which is younger than the guideline-recommended age for initiating routine population-based screening.3,4,11,12 CRC screening recommendations for individuals with LS include screening before the age of 50, possibly as young as age 20–25 or 2–5 years before the earliest diagnosis of cancer if under 25 years of age,6,12,13 and regular transvaginal ultrasound screening for endometrial and ovarian cancer beginning as early as age 30.6,14 In addition, it is recommended that females with LS consider prophylactic hysterectomy with bilateral salpingo-oophorectomy on completion of childbearing.6,13,14 Preliminary evidence also shows that individuals who are diagnosed with LS-associated cancers have a reduced response to fluorouracil chemotherapy treatment.15,16,17 Therefore, identification of LS is important for developing appropriate screening and treatment regimens in affected individuals and for informing and identifying at-risk family members of this disease.
To facilitate identification of individuals with LS, a number of different clinical guidelines have been established. In 2009, the Evaluation of Genetic Applications in Practice and Prevention (EGAPP)7 Working Group recommended universal screening of all patients newly diagnosed with CRC.7 A joint guideline released in 2012 from the National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer (NSGC/CGA-ICC) also supports the EGAPP recommendation and expands it to include all patients newly diagnosed with endometrial cancer,5 on the basis of recent cost-effectiveness studies.18,19,20 By contrast, the current National Comprehensive Cancer Network (NCCN) and American Cancer Society guidelines recommend LS screening once CRC is diagnosed13,21 based on two established criteria that include family history elements, the Amsterdam II criteria,22 and the revised Bethesda criteria.9
A significant gap in the literature concerns how health-care organizations currently screen for LS in individuals with newly diagnosed CRC. Here, we describe LS screening frequency in integrated health-care-delivery settings using a multi-institutional cohort of individuals diagnosed with metastatic CRC between 2004 and 2009. We also examine (i) what, if any, patient factors are most likely to trigger LS screening; (ii) whether physicians routinely collect all of the family history information necessary to assess LS risk using one of the recommended guidelines ( Table 1 ); and (iii) whether the family history information is used to guide LS screening decisions.
Materials and Methods
Study population
The study population included 1,188 patients enrolled at any one of the seven participating Cancer Research Network study institutions23 as part of the Comparative Effectiveness Research in Genomics of Colon Cancer study.24 The seven study institutions are geographically distributed within the United States and represent diverse populations including those of Kaiser Permanente Northwest (Oregon and Washington), Kaiser Permanente Northern California, Kaiser Permanente Colorado, Kaiser Permanente Hawaii, Marshfield Clinic (Wisconsin), Henry Ford Health System (Michigan), and HealthPartners Institute for Education and Research (Minnesota). Eligible patients were identified through tumor registries linked with electronic health information. We included patients aged 18 or older, diagnosed with stage IV CRC between 1 January 2006 and 31 December 2009, and patients diagnosed with stage III CRC between 1 January 2004 and 31 December 2008 who progressed to distant metastatic CRC. Only patients diagnosed with metastatic CRC were included in the study because the primary purpose of the study was to assess questions about treatment that are only indicated for patients with metastatic disease.24 We included only cases diagnosed while in the health plan and excluded cases with incomplete treatment data or without pathology specimens. Because only individuals who were within an insurance health plan were included, all individuals did have insurance and 333 individuals were covered by Medicare/Medicaid.24
This study was approved by the institutional review boards at Kaiser Permanente Northwest, Kaiser Permanente Hawaii, Kaiser Permanente Colorado, Marshfield Clinic Research Foundation, and Henry Ford Health System, and did not require written informed consent. The institutional review boards for the remaining institutions ceded authority to the Kaiser Permanente Northwest institutional review board.
Electronic data collection
Data were extracted from the combined electronic medical records (EMRs) at each institution, using a model of distributed computer code through the Virtual Data Warehouse and the local tumor registry to identify eligible cases and obtain electronic data. The Virtual Data Warehouse is a federated database that defines a standard specification for data contributed by each participating integrated health-care-delivery organization from clinical and administrative data sources.25 Data elements extracted from the Virtual Data Warehouse included tumor registry information, vital signs, procedures, pharmacy, enrollment, and census data.
Manual chart abstraction
We performed manual chart abstraction of EMRs for information that is not typically documented in coded fields but is available from physician notes and other text or scanned documents. Abstracted variables included verification of eligibility, patient characteristics (race, ethnicity, smoking, and alcohol use), a family history of cancer, and cancer treatment history, including surgery, radiation, and chemotherapy, palliative care, and imaging to assess disease progression. Genetic testing variables were manually abstracted to ensure that all records (even external documents) within a patient’s medical record were captured. Any tests ordered by physicians unaffiliated with the health-care organizations or performed by an external laboratory were included, if they were present within a patient’s comprehensive medical record. These variables included genomic microsatellite instability (MSI) and protein immunohistochemistry (IHC) tests, germline mutation analysis of MLH1, MLH2, MLH6, and PMS2 genes, as well as MLH1 hypermethylation studies and BRAF testing to detect sporadic mismatch repair defects.5,11 Family history information was abstracted from coded and text fields, including the clinician notes in the medical chart, and was recorded verbatim and codified in a study database. If a pedigree was available, it was deidentified and included with the chart abstraction.
LS risk and family history analysis
Family history information was coded by a single investigator with a background in genetic counseling (A.K.R.) with assistance from a research assistant (A.Q.L.). Positive family history included any family history of cancer and was further subclassified into a family history of Lynch-associated cancers or non–Lynch associated cancers. Negative family history was coded as “no family history of cancer,” “no family history of colon cancer,” “negative/noncontributory family history,” or “family history unknown” as documented in the medical record. Detailed text-based family history information was recoded for personal and family information about multiple primary colon cancers, additional personal history of other cancers, cancer in first- and second-degree relatives, cancer types, and ages of diagnoses.
We used algorithms, described in Table 1 , to identify individuals with sufficient information eligible for LS screening via Amsterdam II22 or revised Bethesda criteria,9 or MMRPredict26 or PREMM (1,2,6) model.27 Although the NSGC/CGA-ICC, and EGAPP recommendations and the MMRPredict and PREMM models were not available at the time of diagnosis for all members of this cohort, we included them with the cutoff values suggested in the publications26,27 to determine if sufficient information already existed in the medical record to assess individual risk using these guidelines.
Analysis
We compared patient characteristics in individuals selected for LS screening versus those with no LS screening. We defined LS screening to include any guideline-recommended combination of genomic MSI and/or protein IHC tests and/or germline mutation analysis of MLH1, MLH2, MLH6, and PMS2 genes, as well as MLH1 hypermethylation studies and BRAF testing to detect sporadic mismatch repair defects.5,7,11 For individuals with and without documentation of LS screening tests, we compared the number who met the criteria for LS evaluation according to most available personal cancer history and family history–based guidelines ( Table 1 ). All tests were conducted using SAS Release 9 (SAS Institute, Cary, NC). Reported P values were computed using Pearson’s χ2 test (dichotomous variables) or Fisher’s exact tests where cell size was <5 or the two-sample t-test (quantitative variables).
Results
The cohort consisted of 1,188 individuals from seven different institutions; 76% were diagnosed with metastatic CRC and 24% were diagnosed with stage III CRC and had progressed to metastatic disease. The average age at diagnosis was 66.3 years; 74% were non-Hispanic white, and 50.7% were male. Overall, 41 patients (~3% of the sample) were screened for LS via any testing method (MSI, IHC, or germline genetic testing) ( Table 2 ). Individuals with LS screening had a younger age at diagnosis (58 vs. 71 years; P < 0.0001), were more likely to have family history documentation (88 vs. 78%; P = 0.0035), and were more likely to have a positive family history of CRC (22 vs. 6%; P < 0.0001). Screened individuals were also more likely to have documentation of ever using alcohol (68 vs. 43%; P < 0.0001) and less likely to have ever smoked (37 vs. 52%; P < 0.0001).
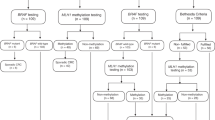
To determine how LS screening guidelines were applied to the 41 individuals screened, we compared the actual tests used with the recommended LS screening guidelines, which indicate screening for either MSI or IHC or both before proceeding to germline sequencing ( Figure 1 ).7 Thirty nine patients (95%) started with MSI or IHC tumor testing. All 13 patients with IHC tests were first negative for LS; however, three individuals (23%) were not tested for all four genes. Of the 14 patients starting with MSI tests, 12 (86%) were MSI low; one was MSI high and had no further testing, and one received MSI testing but lacked test results or follow-up. Of the 12 patients with concurrent MSI and IHC tests, all genes were present on IHC. One was also MSI high and was subsequently found to have the BRAF v600E sporadic mutation.5,11 One patient with MSI-low results also had BRAF testing; however, the BRAF test was conducted a year later, which may indicate testing for antiepidermal growth factor receptor treatment rather than for LS risk. Two patients had selective genetic sequencing only with no mutations found; however, none were tested for all four genes, despite available testing at time of diagnosis ( Figure 1 ).

Flow diagram of Lynch syndrome (LS) screening conducted among patients in the study sample. *LS screening indicates any guideline-recommended combination of genomic MSI and/or protein immunohistochemistry (IHC) tests, germline mutation analysis of MLH1, MLH2, MLH6, and PMS2 genes, and MLH1 hypermethylation studies and BRAF testing to detect sporadic MMR defects (refs. 5,7,11). †Further testing is not indicated when tumors are MSI-L or MSI-S, or when all gene protein products are present on IHC because patients are assumed not to have LS (refs. 4,20,37,38). ‡Further testing is not indicated in the case of MLH1 methylation or BRAF v600E mutation because patients are assumed to not be at risk for LS (refs. 3,5,7). MMR, mismatch repair; MSI-H indicates microsatellite instability high; MSI-L indicates microsatellite instability low; MSI-S indicates microsatellite instability stable.
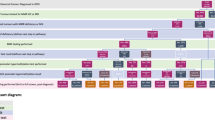
Due to the low rate of LS screening, we investigated documentation of family history (ever documented in record/no documentation found) as the first step toward hereditary cancer-risk assessment. Overall, 937 (79%) patients had documentation of family history, with 719 individuals having sufficient information recorded to evaluate for LS risk using family history–based criteria, after excluding individuals with “family history unknown,” “no family history of colon cancer,” “negative/noncontributory family history,” and individuals with family history recorded in relatives but without a recorded age of diagnosis ( Figure 2 ). Of the 937, 577 (62%) described a family history of any cancer, 350 (37% of 937) were described as negative, and 10 (1% of 937) had family history information that could not be determined. Of patients with a negative family history, 249 (71% of 350) were documented as having “no family history of cancer,” 62 (18% of 350) had “no family history of colon cancer,” and 39 patients (11% of 350) were recorded as having “negative/noncontributory family history” ( Figure 2 ). Because we do not know what the patient was asked in these 39 instances, we cannot accurately assess whether these family histories included any other LS-associated cancers or not.

Availability and completeness of family history information recorded in patient medical records. *Cases of “No family history information recorded” (n = 251) were not evaluated for Lynch syndrome (LS) risk. †Total n = 719 (of 937 with family history documentation) were evaluated for LS risk: “Family history unknown” (n = 10), “No family history of colon cancer” (n = 62), “Negative/noncontributory family history” (n = 39), and “≥1 relative with CRC or Lynch-related cancer AGE NOT RECORDED” (n = 107) could not be evaluated for Lynch syndrome risk due to insufficient information. CRC, colorectal cancer.
Of the 577 patients with a family history of cancer, 186 (32%) noted a history of non–LS-associated cancers only ( Figure 2 ). Of those with a family history positive for CRC or LS-associated cancer, 261 (67% of 391) had sufficient information to determine the patient met the Amsterdam or revised Bethesda criteria for further LS screening ( Figure 2 ). An additional 107 patients (27% of 391) with a family history of CRC or LS-associated cancer could not be accurately assessed for LS risk, usually due to missing age of diagnosis in family members.
Using family history and diagnosis information available in the chart and the algorithms for LS screening guidelines, we also examined the percentage eligible for LS screening versus the percentage actually screened. By any of the nine standards for referral criteria evaluated, the majority of eligible patients were not screened for LS, ranging from 75 (Amsterdam II criteria) to 100% (individuals with multiple CRCs) ( Table 3 ). Of the individuals who did receive LS screening, the majority were appropriate candidates based on the referral criteria evaluated, including the revised Bethesda criteria (28/41 = 68%), the PREMM model (35/41 = 85%), the MMRPredict model (34/41 = 83%), and the EGAPP recommendation or the NSCG/CGA-ICC joint guideline (41/41 = 100%).
Discussion
We found underutilization of LS screening (41/1,188 patients across all sites). When LS screening was conducted, the majority of patients followed the recommended screening cascade. Initial screening for LS can occur through IHC or MSI testing of tumor tissue. Current guidelines agree the most cost-effective process is utilizing tumor testing via IHC and/or MSI first to guide germline genetic testing, with no consensus that one of these tests is better than the other; often both are performed.19,20 Consistent with LS screening guidelines for no preference between MSI and/or IHC as a first step,5,7 we observed approximately equal numbers screened with either test or both tests concurrently. Where the recommended LS screening process was not followed, it was due to incomplete testing of all the genes, lack of follow-through on a positive MSI or IHC test result, or no confirmation of MSI/IHC testing in the chart. Because BRAF testing and other testing such as MSI may be conducted for treatment decisions rather than LS screening, we did investigate whether this may be occurring. In only one instance, did we find a suspicion of BRAF testing that was probably ordered for treatment based on the temporal relationship among diagnosis, testing, and treatment.
We examined LS screening indicators such as family history, multiple primary colon cancers, and age at diagnosis. Lack of family history information does not appear to be the reason for the low rate of LS screening, as 79% of our population had some documentation of family history. To be informative for LS, a family history should document any history of LS-associated cancers beyond CRC and include the cancer type, relative affected (including the lineage), and the age at diagnosis. Like Wood,28 we found sufficient information to assess for LS risk in two-thirds of charts, and 67% of the charts with a positive cancer history included enough information indicating LS risk based on the Amsterdam II or revised Bethesda criteria. Of those individuals we could not assess, the charts frequently lacked age at diagnosis information for relatives with a LS-associated cancer. This observed lack of complete family history information is also consistent with other reports.29,30,31 Several other studies have also found that insufficient family history information in the EMR was the limitation for implementation of the Bethesda and Amsterdam II criteria.32,33 Singh and colleagues33 found that there was no documentation of family history in 7.6% of the records within the tertiary care EMR at the VA hospital, with age at cancer onset incomplete in up to 49% of the population. Similarly, there was incomplete documentation within the EMR of the Penn State Hershey Cancer Institute in 38% of the patients diagnosed with an LS-associated cancer within the study period.32
We were able to determine that ~22% of our study population was eligible for LS screening using the revised Bethesda criteria;9 1% of the study population fell into the more restrictive Amsterdam II criteria.22 Yet only 3% of the population had any LS screening. This is consistent with multiple studies reporting underutilization of screening and referrals,29,30,32,33,34,35 ranging from 7% to 9% of study populations receiving LS screening. We determined that 89% of patients meeting the Bethesda criteria and 91% of patients with a positive family history of CRC or LS-associated cancer in more than two relatives did not have LS screening, despite their elevated risk status. Despite the availability of LS screening programs at these organizations, no cases of LS were identified, and the only patient with a MSI-high result did not receive further testing. If we consider that 3% of CRCs would have had LS,3,4,6,7,11 then ~35 cases of LS were missed among the 1,188 members in the study population.
To improve LS identification, there is clearly a need for reflexive (automatic) or universal screening of all newly diagnosed patients with CRC. Beamer and colleagues36 determined that 71% of the National Cancer Institute Comprehensive Cancer Centers were conducting reflexive LS screening, whereas only 15% of the community hospital cancer programs regularly screened for LS. Similar to our results, there was no consensus on initial screening test method, with 48% of programs testing via IHC, 14% using MSI, and 38% using both screening tests. Screening at these institutions was rarely universal and was often triggered by particular criteria such as young age at onset, especially in the community hospital cancer programs. However, it is estimated that one in four individuals with LS will be missed by the use of family history or age-based screening criteria.37 Recently, Moreira and colleagues38 used pooled data from four large cohorts of newly diagnosed patients with CRC to determine that universal mismatch repair testing of newly diagnosed patients with CRC had greater sensitivity to detect LS as compared with all other strategies, further strengthening the argument for universal screening. None of the institutions in our study were Comprehensive Cancer Centers, and none had institutional screening guidelines nor did they have universal screening programs at the time of the study; however, several organizations are currently taking steps to either implement or evaluate such programs (K. Goddard, personal communication).
Our study has several limitations. We examined only metastatic CRC cases in this analysis. Providers may not consider LS screening in patients with advanced cancer due to decreased relevance of recurrence risk and additional cancers in the patient. However, investigation of LS screening within three of the institutions did not show a difference in LS screening for earlier stage at diagnosis (data not shown). We may have also underestimated the number of patients eligible for LS screening because “negative” or “noncontributory” family history does not necessarily mean enough information was asked to determine LS risk, and “no family history of colon cancer” may indicate that family history of endometrial and other LS-associated cancer was not assessed. Although we captured any cancer history for family members, for study participants, we collected information only on multiple primary CRCs and did not assess whether any individuals had a previous diagnosis of other LS-associated cancers; therefore, our estimate of eligible individuals due to multiple LS-associated cancers may also be low.
In summary, we found very low identification and screening of newly diagnosed cancer patients for LS. Family history of CRC is documented for most individuals, although this varies by site. Despite the brevity with which family history is recorded, a significant proportion of patients in the study population had enough family history information recorded to assess risk status for LS. However, only 3% of patients meeting criteria were screened for LS, representing severe underdiagnosis of this hereditary condition in patients and their families.
Disclosure
The authors declare no conflict of interest.
References
American Cancer Society. Cancer facts and figures 2012. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-031941pdf. Accessed 29 July 2012.
US Cancer Statistics Working Group, US Department of Health and Human Services, Centers for Disease Control and Prevention, National Cancer Institute. United States Cancer Statistics: 1999–2008 incidence and mortality web-based report, 2012 update. http://apps.nccd.cdc.gov/uscs. Accessed 19 December 2012.
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851–1860.
Lynch HT, de la Chapelle A . Hereditary colorectal cancer. N Engl J Med 2003;348:919–932.
Weissman SM, Burt R, Church J, et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. J Genet Couns 2012;21:484–493.
Weissman SM, Bellcross C, Bittner CC, et al. Genetic counseling considerations in the evaluation of families for Lynch syndrome–a review. J Genet Couns 2011;20:5–19.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009;11:35–41.
Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol 2012;30:958–964.
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261–268.
Bonadona V, Bonaïti B, Olschwang S, et al.; French Cancer Genetics Network. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305:2304–2310.
Bellcross CA, Bedrosian SR, Daniels E, et al. Implementing screening for Lynch syndrome among patients with newly diagnosed colorectal cancer: summary of a public health/clinical collaborative meeting. Genet Med 2012;14:152–162.
Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM ; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009;104:739–750.
National Comprehensive Cancer Network. Clinical practice guidelines in oncology: Colorectal cancer screening version 3.2012. http://www.ncc.org. Accessed 2 August 2012.
Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006;296:1507–1517.
Guastadisegni C, Colafranceschi M, Ottini L, Dogliotti E . Microsatellite instability as a marker of prognosis and response to therapy: a meta-analysis of colorectal cancer survival data. Eur J Cancer 2010;46:2788–2798.
Sinicrope FA, Foster NR, Thibodeau SN, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst 2011;103:863–875.
Des Guetz G, Schischmanoff O, Nicolas P, Perret GY, Morere JF, Uzzan B . Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur J Cancer 2009;45:1890–1896.
Mvundura M, Grosse SD, Hampel H, Palomaki GE . The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med 2010;12:93–104.
Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a cost-effectiveness analysis. Ann Intern Med 2011;155:69–79.
Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN . EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 2009;11:42–65.
Levin B, Lieberman DA, McFarland B, et al.; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008;134:1570–1595.
Vasen HF, Watson P, Mecklin JP, Lynch HT . New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999;116:1453–1456.
The HMO Cancer Research Network. The HMO Cancer Research Network: Capacity, Collaboration, & Investigation. NIH Publication No. 10-6448 ed. US Department of Health and Human Services: Washington, DC, 2010.
Webster J, Kauffman TL, Feigelson HS, et al.; CERGEN study team. KRAS testing and epidermal growth factor receptor inhibitor treatment for colorectal cancer in community settings. Cancer Epidemiol Biomarkers Prev 2013;22:91–101.
Hornbrook MC, Hart G, Ellis JL, et al. Building a virtual cancer research organization. J Natl Cancer Inst Monographs 2005;35:12–25.
Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 2006;354:2751–2763.
Kastrinos F, Steyerberg EW, Mercado R, et al. The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology 2011;140:73–81.
Wood M, Kadlubek P, Lu KH et al. Quality of cancer family history and referral for genetic counseling and testing among oncology practices: a pilot test of quality measures as part of the ASCO Quality Oncology Practice Initiative (QOPI) [abstract]. J Clin Oncol 2012;30:Abstr CRA1505.
Grover S, Stoffel EM, Bussone L, Tschoegl E, Syngal S . Physician assessment of family cancer history and referral for genetic evaluation in colorectal cancer patients. Clin Gastroenterol Hepatol 2004;2:813–819.
Murff HJ, Greevy RA, Syngal S . The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007;10:174–180.
van Dijk DA, Oostindiër MJ, Kloosterman-Boele WM, Krijnen P, Vasen HF ; Hereditary Tumor Study Group of the Comprehensive Cancer Centre West. Family history is neglected in the work-up of patients with colorectal cancer: a quality assessment using cancer registry data. Fam Cancer 2007;6:131–134.
Mukherjee A, McGarrity TJ, Ruggiero F, et al. The revised Bethesda guidelines: extent of utilization in a university hospital medical center with a cancer genetics program. Hered Cancer Clin Pract 2010;8:9.
Singh H, Nugent Z, Demers AA, Bernstein CN . Rate and predictors of early/missed colorectal cancers after colonoscopy in Manitoba: a population-based study. Am J Gastroenterol 2010;105:2588–2596.
Tranø G, Wasmuth HH, Sjursen W, Hofsli E, Vatten LJ . Awareness of heredity in colorectal cancer patients is insufficient among clinicians: a Norwegian population-based study. Colorectal Dis 2009;11:456–461.
Van Lier MG, De Wilt JH, Wagemakers JJ, et al. Underutilization of microsatellite instability analysis in colorectal cancer patients at high risk for Lynch syndrome. Scand J Gastroenterol 2009;44:600–604.
Beamer LC, Grant ML, Espenschied CR, et al. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol 2012;30:1058–1063.
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26:5783–5788.
Moreira L, Balaguer F, Lindor N, et al.; EPICOLON Consortium. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012;308:1555–1565.
Acknowledgements
This research was conducted at multiple institutions of the Health Maintenance Organization (HMO) Cancer Research Network (CRN). The CRN consists of the research programs, enrollee populations, and databases of 14 members of the HMO Research Network. The overall goal of the CRN is to conduct collaborative research to determine the effectiveness of preventive, curative, and supportive interventions for major cancers that span the natural history of those cancers among diverse populations and health systems. The 14 health plans, with nearly 11 million enrollees, are distinguished by their longstanding commitment to prevention and research, and collaboration among themselves and with affiliated academic institutions. The Comparative Effectiveness Research in Genomics of Colon Cancer Project Team includes the following institutions and individuals: Henry Ford Health System: G. Alexander, R., Faryniarz, J. Garhy, C. Hwang, R. Krajenta, P. Meier; Health Partners Institute for Research and Education: C. Anderson, A. Behl, T. Defor, T. Flottemesch, M. Maciosek, A. Nelson, B. Owens, P. Pawloski, L. VanArman; Kaiser Permanente Northwest: K. Bennett, P. Dandamudi, K. Goddard, R. Holmes, S. Houston, T. Kauffman, S. Lai, J. Lin, C. Machen, R. Meenan, J. Mesa, K. Muessig, M. O’Keeffe-Rosetti, K. Olson, D. Schwarzkopf, C. Senger, N. Tirumali, C. Wax, E. Webber, J. Webster, E. Whitlock, S. Zuber; Kaiser Permanente Hawaii: K. Bow, J. Carney, Y. Daida, S. Honda, A. Stone-Murai, A. Vinoya, A. Williams, C. Wong; Department of Public Health Colorado: N. Colange; Marshfield Clinic Research Foundation: C. Cold, D. Cross, J. Engel, T. Kitchner, A. Onitilo; Kaiser Permanente Georgia: R. Davis, M. Hemmes; Kaiser Permanente Colorado: T. Delate, H. Feigelson, J. Freml, K. Funk, A. Le, J. McCance, A. Menter, C. Nyirenda, A. Rahm, C. Zeng; Kaiser Permanente Northern California: M. Ford, J. Harris, K. Huang, S-F. Jiang, T. Kolevska, L. Kushi, P. Liljestrand, A. Mirabedi, J. Morse, C. Somkin. University of Hawaii: L. LeMarchand; University of Washington: D. Veenstra; Oregon Health and Sciences University: M. Johnson, C.S. Richards; Georgetown University: S. O’Neill; Essentia Institute of Rural Health: C. McCarty, P. Hitz; This study was funded by a grant from the National Cancer Institute 5UC2CA148471 (Principal Investigators: K. Goddard, L. Kushi, and E. Whitlock).
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
About this article
Cite this article
Cross, D., Rahm, A., Kauffman, T. et al. Underutilization of Lynch syndrome screening in a multisite study of patients with colorectal cancer. Genet Med 15, 933–940 (2013). https://doi.org/10.1038/gim.2013.43
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.43
Keywords
This article is cited by
-
Identifying patients with Lynch syndrome using a universal tumor screening program in an integrated healthcare system
Hereditary Cancer in Clinical Practice (2022)
-
Literacy-adapted, electronic family history assessment for genetics referral in primary care: patient user insights from qualitative interviews
Hereditary Cancer in Clinical Practice (2022)
-
Room for improvement: One third of Lynch syndrome patients presenting for genetic testing in a highly specialised centre in Stockholm already have cancer
Hereditary Cancer in Clinical Practice (2021)
-
Advances in Hereditary Colorectal Cancer: Opportunities and Challenges for Clinical Translation
Current Genetic Medicine Reports (2020)
-
Underutilization of Lynch Syndrome Screening at Two Large Veterans Affairs Medical Centers
Digestive Diseases and Sciences (2020)
