
Abstract
Keratoglobus is a rare noninflammatory corneal thinning disorder characterised by generalised thinning and globular protrusion of the cornea. It was first described as a separate clinical entity by Verrey in 1947. Both congenital and acquired forms have been shown to occur, and may be associated with various other ocular and systemic syndromes including the connective tissue disorders. Similarities have been found with other noninflammatory thinning disorders like keratoconus that has given rise to hypotheses about the aetiopathogenesis. However, the exact genetics and pathogenesis are still unclear. Clinical presentation is characterised by progressive diminution resulting from irregular corneal topography with increased corneal fragility due to extreme thinning. Conservative and surgical management for visual rehabilitation and improved tectonic stability have been described, but remains challenging. In the absence of a definitive standard procedure for management of this disorder, various surgical procedures have been attempted in order to overcome the difficulties. This article reviews the aetiological factors, differential diagnosis, histopathology, and management options of keratoglobus.
Similar content being viewed by others
Introduction
The noninflammatory corneal ectasia are a group of disorders characterised by corneal thinning, protrusion, and scarring. Keratoglobus forms a rarer subset of this group.1 In the past, it was considered synonymous with megalocornea and congenital glaucoma. However, in 1947, Verrey2, through detailed descriptions of his patients, was able to show that it was a distinct clinical entity. This was further supported by Cavara3 in 1950. The exact cause remains largely unknown although various theories have been proposed based on its similarities with other more common noninflammatory ectasia such as keratoconus. In fact, these similarities have brought about confusion as to whether the disorders comprising this group are separate clinical disorders, or rather a spectrum of the same disease process.
Aetiological factors
Keratoglobus is primarily considered a congenital disorder present since birth.3, 4, 5 However, in more recent years, there have been reports of acquired forms of keratoglobus. The congenital form of the disorder is always bilateral. The exact genetics of the disorder have not been studied in detail and no definite inheritance pattern has been described. It is assumed to be autosomal recessive, as described by Poliquen et al.4, 5 It has also been associated with disorders of the connective tissue such as Ehlers–Danlos syndrome, Marfan syndrome, and Rubinstein–Taybi syndrome (Table 1).6 Initially, there were reports of keratoglobus in relation to ‘blue sclerae’.5, 7, 8 These blue sclera syndromes were actually thought to be manifestations of the aforementioned syndromes, including osteogenesis imperfecta.9, 10 The seemingly blue sclera is caused by a thinned and more transparent sclera, maximally at the ciliary body. Ehlers–Danlos syndrome type VI, in particular, is distinct for its ocular manifestations.11 These include corneal abnormalities of cornea plana, keratoconus and keratoglobus, blue sclera, and ocular fragility. Other systemic features in this type are the presence of hypermobile joints, skeletal abnormalities like scoliosis, pectus excavatum, a marfanoid habitus, and hearing loss. Skin laxity and fragility is not a characteristic finding, unlike in other types of Ehlers–Danlos syndromes, and lysyl hydroxylase activity may be normal.11, 12 Keratoglobus has also been described in cases of Leber’s congenital amaurosis.13
The acquired forms of keratoglobus have been described in association with vernal keratoconjunctivitis, chronic marginal blepharitis, idiopathic orbital inflammation,14 and dysthyroid eye disease.15 In the case of vernal keratoconjunctivitis and chronic marginal blepharitis, the corneal ectasia may be related to frequent eye-rubbing.14, 16 This has been thought by some authors to be a factor in the development of keratoconus, although the exact mechanism of such an association has not been proven.17 Overlaps in aetiological factors between keratoconus, pellucid marginal degeneration, and keratoglobus, such as their manifestations in connective tissue disorders and various acquired forms has further fuelled the speculation about their being different spectrums of the same disease. Also, there are reports of keratoconus and keratoglobus,3, 14, 18 as well as pellucid marginal corneal degeneration (PMCD) and keratoglobus19, 20 being clinically documented in the same patient over time. Poliquen et al4 classified keratoglobus into two types, one being congenital and the other acquired. He described the acquired type as being a severe form of keratoconus. Topographical analysis of pellucid marginal degeneration and keratoglobus by Karabatsas and Cook19 includes a case report of a patient diagnosed with pellucid marginal degeneration in one eye and who was later found to have developed a keratoglobus-like picture on the follow-up. This led them to hypothesise that the natural history of pellucid marginal degeneration may be in the development of keratoglobus by circumferential extension of the peripheral gutter. Similarly, Cameron14 described a case report documenting kerotoconus progressing into a keratoglobus-like picture. However, a constant association and progression between these ectatic disorders has not been described to validate these hypotheses. In the reports of keratoglobus associated with thyroid ophthalmopathy15 and orbital inflammatory disease,14 the authors hypothesise ischaemia of the anterior segment secondary to these conditions resulting in diffuse, progressive ectasia of the cornea. Case reports of keratoglobus with syphilis, a post-traumatic case and posterior polymorphous dystrophy have also been described in the literature.18, 21
Clinical findings
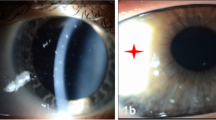
Keratoglobus is a bilateral ectatic disorder of the cornea, principally characterised by a globular protrusion of the cornea associated with diffuse thinning from limbus to limbus. The age of onset is at birth. The thinning is commonly maximal at the periphery and may be up to one-fifth the normal corneal thickness (Figure 1a). Other corneal parameters, however, are normal, including a normal corneal diameter that is an important criterion in differentiating it from conditions such as buphthalmos. Patients generally present with clear corneas unless they undergo acute episodes of hydrops and scarring. Vogt striae and Fleischer’s rings are not associated with keratoglobus.1, 14, 18

Slit-lamp photograph of a patient with bilateral keratoglobus (a) showing thinning (maximum at the periphery) and bulging of cornea, (b) after ‘tuck-in’ lamellar keratoplasty (TILK).
As a result of the thinning and protrusion, there is high myopia with irregular astigmatism, which is the main cause of poor vision in these patients, and is difficult to treat with refractive correction. Other than poor vision patients are generally asymptomatic. However, owing to extreme thinning and fragility of the cornea, many cases may initially present with corneal perforations, either spontaneous or following minimal trauma.7, 10, 14, 18 In such cases, the diagnosis of keratoglobus must be kept in mind. This is especially important as surgical closure in these cases is difficult because of the extreme thinness of the cornea that leads to cut through of the sutures and inability for wound closure. As mentioned earlier, spontaneous tears in Descemet’s membrane may occur resulting in acute presentations of pain, tearing, photophobia, and sudden diminution of vision of acute hydrops.14, 22, 23 The loss of endothelial cell barrier leads to fluid accumulation within the stroma. Resolution may take months, with variations from 5 to 36 weeks reported in other corneal ectatic diseases. A study of anterior segment optical coherence tomography of cases involving acute hydrops described two stages in resolution.24 The first was reattachment of Descemet’s membrane, followed by endothelial migration across the tear. There are no other associated ocular abnormalities with keratoglobus.
The diagnosis of keratoglobus is essentially a clinical one owing to the characteristic clinical findings. Confusion might arise in less severe cases where there may be difficulty in differentiating the condition from other ectatic conditions. Investigational modalities include ultrasonic pachymetry that would demonstrate reduced corneal thickness and corneal topography by Orbscan TECHNOLAS Perfect Vision GmbH (Munich, Germany) that would show diffuse thinning.17, 19, 25, 26 However, these investigational modalities may not be possible to carry out in cases of more severe corneal distortion. There is limited literature describing exact topographical pictures of keratoglobus. A description by Karabatsas and Cook19 of a patient with both pellucid marginal degeneration and keratoglobus described the videokeratography image, showing irregular astigmatism with irregular power distribution of the eye with keratoglobus. There was a peripheral arc of increased power or steepening resulting in flattening of ‘arching’ of the bow tie configuration of topography. They felt that the peripheral arc of increased power reflected a progression of PMCD by extension of the inferior peripheral thinning circumferentially.19 Another case report of keratoglobus in association with posterior polymorphous dystrophy showed Orbscan findings of generalised steepening of both anterior and posterior curvatures, with irregular astigmatism and asymmetric bow tie pattern.21
Systemic evaluation might point towards connective tissue disorder. These include features mentioned earlier of blue sclera, joint hypermobility, skeletal abnormalities, hearing loss, abnormal dentition, high-arched palate, as well as other features specific to each syndrome. It has been noted in few case series that keratoglobus in association with blue sclera syndromes is more probable to undergo spontaneous perforation or after minimal trauma, and hence the name ‘brittle cornea’.7, 14
Histopathology
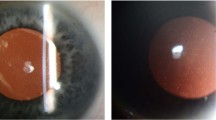
The anatomical features that have been described for keratoglobus corneas are frequent disruptions or complete absence of Bowman’s layer, stromal thinning and disorganisation, and breaks or thickening of Descemet’s membrane (Figure 2).4, 14, 27 Stromal thinning is most marked at the periphery or mid-periphery, as seen clinically. Poliquin et al4 described different histopathological findings for the acquired type of keratoglobus, which is characterised by an essentially normal Bowman’s layer that undergoes focal breaks with superficial stromal ectasia secondary to other corneal pathology. The histopathalogical findings are similar to that of keratoconus.

Photomicrograph of (a) lamellar corneal button showing diffuse stromal thinning (stain: haematoxylin and eosin, × 40), (b) central cornea showing thinned corneal epithelium with intraepithelial oedema and separation of epithelium from epithelial basement membrane. There is discontinuity of Bowman’s layer (marked between the two red arrows and two black arrows; stain: haematoxylin and eosin, × 400).
Immunohistochemical studies of keratoconus have been done, but are limited in cases of keratoglobus. A study done by Meghpara et al27 described the immunohistochemical features of nine corneal buttons of keratoglobus patients in comparison with keratoconus and normal cornea. They found decreased expression of proteinase inhibitor alpha-1-PI, and increased expression of the transcription factor Sp1 in the corneal epithelial cells. This imbalance leads to an alteration in tissue degradation processes within the cornea. In addition, they found increased expression of matrix metalloproteinases (MMPs) 1, 2, and 3 within the epithelial cells. The MMPs are responsible for the degradation of some component of the extracellular matrix and are transcriptionally upregulated by various inflammatory mediators and inhibited by tissue inhibitors of metalloproteinases.28 Their role in wound repair and remodelling is well known.29 Similar findings were observed in keratoconus. However, in keratoconus, there was an increased expression of only MMP 1, which was maximal at the centre, corresponding to the area of maximal thinning in this disease. In comparison, increased Sp1 and MMP 1, 2, and 3 expression was found diffusely throughout the cornea, but maximally at the mid-periphery and corresponded to areas of underlying Bowman’s layer disruptions. The increased expression of pro-degradation products and decreased expression of inhibitory substances is most probably a key pathogenic factor in causing ectasia.
Differential diagnosis
The main differential diagnosis of a case of keratoglobus is the other noninflammatory ectatic disorders. These include keratoconus, pellucid marginal degeneration, and posterior keratoconus.1, 17, 29 In addition, confusion might arise in young patients in differentiating keratoglobus from congenital glaucoma and megalocornea.
In a case of megalocornea, the main differentiating feature is the increased corneal diameter (usually over 12.5 mm) with absence of any corneal thinning. This is in contrast to keratoglobus where corneal diameters are normal and there is profound, diffuse thinning. There is therefore an absence of any corneal protrusion, astigmatism, hydrops, or scarring in cases of megalocornea. Congenital glaucoma may also present with moderate protrusion of the cornea, hydrops, and mild astigmatism with myopia. However, the hallmark would be raised intraocular pressure and possible glaucomatous optic nerve changes that would be absent in the case of keratoglobus. There is also no corneal thinning and corneal diameters may be increased. In congenital glaucoma, the myopia would result primarily from the increased anterior and posterior axial length, whereas in keratoglobus it would be mainly because of the increased corneal curvature.
Keratoconus is different from keratoglobus in the age of presentation. Whereas keratoglobus presents at birth, keratoconus develops around puberty and may progress until 40–50 years of age. Keratoglobus is considered a non-progressive or minimally progressive disorder. The corneal thinning in keratoconus is most commonly seen in the inferior paracentral aspect of the cornea. The protrusion is commonly described as conical in shape, with maximal thinning at the apex. This is in contradiction to keratoglobus that presents with diffuse thinning and a globular protrusion. Scarring is more commonly found in keratoconus, and there are the presence of Vogt’s striae and Fleischer’s ring. In case of pellucid marginal degeneration, the age of presentation is around 20–40 years. Thinning involves the inferior aspect of cornea, as a band of 1–2 mm width and extending from 4 to 8 o’clock. Protrusion occurs superior to this area of thinning leading to characteristic topographical patterns. Scarring and hydrops may also occur.
Treatment
Treatment of keratoglobus remains challenging to date. Conservative therapy is refractive correction for high myopia, but is limited by the high irregular astigmatism. Use of contact lens and newer scleral lenses have been described, but is still a matter of debate because of the theoretical risk of perforation on contact lens insertion and removal over corneas that are known to perforate on even trivial trauma.14 There are no specific studies or literature on contact lens fitting in cases of keratoglubus. The extreme protrusion and irregularity makes fitting complicated, with a need for balance between optical and lens stability. Customised fitting, on patient to patient basis, of sclera lenses, small diameter rigid gas permeable (RGP) lenses, reverse geometry hydrogel lenses, as well as large diameter inverse geometry RGP lenses have been described for corneal ectasias.30, 31, 32, 33, 34, 35
Of prime importance for these patients is counselling for use of protective eye wear, and avoidance of contact sports owing to the high risk of perforation. However, in children, enforcement of use of protective glasses is difficult making them susceptible to injury.
Treatment of acute hydrops can be both conservative and surgical. Conventional treatment is nonspecific, and involves the use of patching, bandage contact lens, topical hypertonic saline, and cycloplegics to reduce the oedema.22, 23 In recent years, reports on faster resolution with use of intracameral gas had brought a shift towards surgical intervention of acute hydrops. Use of air, sulphur hexafluoride, and perfluoropropane has been described.36, 37, 38, 39, 40, 41 The gas is thought to act as a mechanical barrier to fluid entry into the corneal stroma, as well as a tamponading agent in re-opposing the rolled, detached edges of the Descemet’s tear. This is theoretically felt to aid in endothelial migration across the tear.39 Various studies have shown a significantly faster resolution time with use of these agents as compared with conventional therapy in acute hydrops with keratoconus as well as a few case reports in eyes of kerotoglobus.36, 40, 41 A study by Basu et al41 found that the resolution time did not show much difference between conventional and intracameral perfluoropropane treatment in cases of pellucid marginal degeneration and keratoglobus. They felt that this may have been due to location of breaks in these cases as well as the usually larger, more extensive breaks associated with keratoglobus.
Surgically, there is no known standard procedure for management of the condition owing to its rarity, and therefore scarcity of reports of consistent surgical results. The aim of surgical intervention was earlier restricted to the repair of perforations. As mentioned earlier, this commonly resulted in poor outcomes because of the nature of the perforations that are usually large and stellate, and owing to the fragility of the thinned cornea that prevented stable placement of sutures that would cut through or ‘cheese-wire’.14 Attempts to overcome these problems have been reported in few case reports using intracameral air or perfluoropropane as tamponading agents to achieve wound closure in adjunct to suture closure.42
Conventional penetrating keratoplasty in these patients is also not possible because of the thinned cornea and peripheral graft–host thickness disparity that prevents adequate wound closure. Patients also are left with extreme irregular astigmatism and ultimately poor visual outcome or tectonic stability. Attempts to overcome these problems have led to reports of modifications of the penetrating keratoplasty procedure (Table 2). These include the use of large limbus to limbus donor corneal grafts to avoid placement of the graft–host junction at the thinned mid-periphery, thereby creating better stability. However, this led to loss of the immunological privelege within the avascular cornea, increasing chances of graft rejection, and therefore requiring long-term immunosuppression and its antecedent complications.43, 44 There is also a case report on the use of midsize (9 mm diameter), eccentrically placed, paracentral graft with the aim of centralising the graft at the point of maximum thinning of host cornea in order to avoid the graft–host junction at this point.45
Lamellar keratoplasties have also been attempted in these cases, namely epikeratoplasty. This is a type of onlay lamellar procedure. Following posterior dissection of the conjunctiva and removal of host epithelium, a donor corneoscleral lenticule devoid of endothelium and Descemet’s membrane is placed over the host cornea and sutured over it at the periphery to sclera.9 Epikeratoplasty was first described by Kaufman and Werblin46 in treatment of aphakia, myopia, and keratoconus in children. It is considered a safe and easy procedure, especially in eyes with thinned corneas making extensive host corneal manipulation risky. However, such large onlay grafts disrupt and overlie the host limbal stem cells that results in delayed re-epithelialisation of the graft or persistent epithelial defects that are the commonest complications of the procedure.47, 48 This in turn may lead to increased chances of infection and the ensuing complications. Javadi et al49 have described a variation in the technique to avoid limbal stem cell damage. They described formation of a 360 degree peripheral lamellar intrastromal pocket in the host cornea into which the donor corneal lenticule is inserted peripherally, thereby avoiding any manipulation at the host limbus. Other complications reported include interface opacities and intraepithelial cysts that would affect the final visual outcome.50 However, the procedure provides a stable host bed for a secondary penetrating keratoplasty for visual rehabilitation, should it be required. Such a procedure has been described by Macsai et al,51 and Jones and Kirkness.52 Surgical stability and visual outcome reported though are much better as compared with a conventional penetrating keratoplasty in these eyes.
Another procedure described for surgical management of keratoglobus is corneoscleroplasty, modified to avoid the host angle structures.53 This involves a central full thickness penetrating keratoplasty with a lamellar peripheral corneoscleral dissection from the edge of the full thickness keratoplasty to ∼14 mm. The deeper tissue of the corneoscleral graft is dissected appropriately in the periphery to fit over the lamellar host sclera dissection peripherally. The authors of this procedure report both a good structural integrity and visual outcome in two patients of ectatic corneal disorders with avoidance of secondary glaucoma. However, delayed epithelialisation and immunological graft reactions were complications encountered with the procedure necessitating long-term immunosuppression.
In attempts to overcome the problems in surgical management, many modifications in technique have been described by various authors. Another such procedure described by Vajpayee et al54 is ‘tuck-in’ lamellar keratoplasty. This involves lamellar dissection of the central cornea and lamellar dissection of intrastromal pockets in the periphery into which the peripheral flange of the donor corneal lenticule is tucked in (Figure 1b). The tucked in peripheral flange especially provides structural stabilisation at the point of maximal thinning. Lamellar dissection of an already thinned cornea is technically demanding, however, with chances of perforation during the procedure. Interface opacities associated with lamellar procedures might also be expected, but similar to epikeratoplasty, the initial procedure stabilises the host bed for a future penetrating keratoplasty, should it be required. A mean follow-up of 1.7 years postoperatively in 12 patients undergoing this procedure by Kaushal et al55 showed improvement in best-corrected visual acuity, with significant reduction in the spherical equivalent and astigmatism. There were no cases of interface haze either. The same procedure has been used in both pellucid marginal degeneration and keratoconus by the authors, limiting the peripheral flange to the inferior 180 degrees where maximal thinning is present.
In 2005, Kanellopulous et al56 reported use of a corneoscleral rim over the thinned corneal periphery of a patient with keratoglobus that acted as a buttress while avoiding any manipulation of the central visual area. They felt that this technique slowed the progressive expansion of the thinned mid-periphery, was easy and safe to perform, with minimal chances of immunological reaction, and helped to delay the need for further surgical intervention.
More recently, pentacam-based big bubble deep anterior lamellar keratoplasty was carried out in a series of 50 patients, one of which was a case of keratoglobus.57 Use of pentacam provides a three-dimensional image or thickness profile of the entire cornea preoperatively, which is crucial in the assessment of depth of lamellar trephination for better outcome. However, deep dissection to Descemet’s membrane level is surgically demanding and difficult, more so in ectatic conditions. They report an overall conversion rate of penetrating keratoplasty of 16% using this technique. Details on a case to case basis, such as for the single case of keratoglobus, is not reported.
There is no standard surgical procedure in the management of keratoglobus. Reports of surgical results are limited by the rarity of the condition. Individual procedures have their own advantages and disadvantages, and choice of procedure would depend on individual surgeons’ choice and technical ability.
Conclusion
Keratoglobus is a rare noninflammatory ectatic corneal disorder with limited literature on the subject. Various case reports and case series by different authors have brought out the fact that it is a distinct corneal disorder with characteristic clinical findings and aetiological associations. Although primarily congenital, an acquired form has also been recognised secondary to other corneal pathology, giving rise to a keratoglobus-like picture. Histological and immunohistochemical studies have shown a degradative process causing stromal thinning in both keratoglobus and keratoconus, although the exact pathogenesis is still unclear. This further emphasises the possible connection between different noninflammatory ectatic conditions, leading some authors to believe they are phenotypic variations of the same underlying condition. Treatment remains a challenge both in visual rehabilitation as well as maintaining structural integrity of the cornea. Various surgical procedures have been described, but no treatment standard exists because of limited patient reports and follow-up of newer surgical procedures.
References
Feder RS, Kshettry P . Noninflammatory ectatic disorders. In: Krachmer JH, Mannis MJ, Holland EJ (eds) Cornea: Fundamentals, diagnosis and management 3rd Vol. Elsevier Mosby: New York, 2005 pp 955–974.
Verrey F . Keratoglobe aigu. Ophthalmologica 1947; 114: 284–288.
Cavara V . Keratoglobus and keratoconus: a contribution to the nosological interpretation of keratoglobus. Br J Ophthalmol 1950; 34: 621–626.
Pouliquen Y, Dhermy P, Espinasse MA, Savoldelli M . Keratoglobus. J Fr Ophthalmol 1985; 8 (1): 43–45.
Gregoratos ND, Bartsocas CS, Papas K . Blue sclerae with keratoglobus and brittle cornea. Br J Ophthalmol 1971; 55 (6): 424–426.
Nelson ME, Talbot JF . Keratoglobus in Rubinstein-Taybi syndrome. Br J Ophthalmol 1989; 73 (5): 385–387.
Biglan AW, Brown SI, Johnson BL . Keratoglobus and blue sclera. Am J Ophthalmol 1977; 83 (2): 225–233.
Arkin W . Blue scleras with keratoglobus. Am J Ophthalmol 1964; 58: 678–682.
Cameron JA, Cotler JB, Risco JM, Alvarez H . Epikeratoplasty for keratoglobus associated with blue sclera. Ophthalmology 1991; 98 (4): 446–452.
Hyams SW, Kar H, Neumann E . Blue sclerae and keratoglobus. Ocular signs of a systemic connective tissue disorder. Br J Ophthalmol 1969; 53 (1): 53–58.
Cameron JA . Corneal abnormalities in Ehlers-Danlos syndrome type VI. Cornea 1993; 12 (1): 54–59.
Judisch GF, Waziri M, Krachmer JH . Ocular Ehlers-Danlos syndrome with normal lysyl hydroxylase activity. Arch Ophthalmol 1976; 94 (9): 1489–1491.
Koenekoop RK . An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol 2004; 49 (4): 379–398.
Cameron JA . Keratoglobus. Cornea 1993; 12 (2): 124–130.
Jacobs DS, Green WR, Maumenee AE . Acquired keratoglobus. Am J Ophthalmol 1974; 77 (3): 393–399.
Cameron JA, Al-Rajhi AA, Badr IA . Corneal ectasia in vernal keratoconjunctivitis. Ophthalmology 1989; 96 (11): 1615–1623.
Rabinowitz YS . Keratoconus. Surv Ophthalmol 1998; 42 (4): 297–319.
Baillif S, Garweg JG, Grange JD, Burillon C, Kodjikian L . Keratoglobus: review of the literature. J Fr Ophthalmol 2005; 28 (10): 1145–1149.
Karabatsas CH, Cook SD . Topographic analysis in pellucid marginal corneal degeneration and keratoglobus. Eye 1996; 10 (4): 451–455.
Rumelt S, Rehamy U . Surgically induced keratoglobus in pellucid marginal degeneration. Eye 1998; 12 (1): 156–158.
Harissi-Dagher M, Dana MR, Jurkunas UV . Keratoglubus in association with posterior polymorphous dystrophy. Cornea 2007; 26 (10): 1288–1291.
Gupta VP, Jain RK, Angra SK . Acute hydrops in keratoglobus with vernal keratoconjunctivitis. Indian J Ophthalmol 1985; 33 (2): 121–123.
Grewal S, Laibson PR, Cohen EJ, Rapuano CJ . Acute hydrops in the corneal ectasias: associated factors and outcomes. Trans Am Ophthalmol Soc 1999; 97: 187–198.
Basu S, Vaddavalli PK, Vemuganti GK, Ali MH, Murthy SI . Anterior segment optical coherence tomography features of acute corneal hydrops. Cornea 2012; 31 (5): 479–485.
Lee BW, Jurkunas UV, Harissi-Dagher M, Poothullil AM, Tobaigy FM, Azar DT . Ectatic disorders associated with claw-shaped pattern on corneal topography. Am J Ophthalmol 2007; 144 (1): 154–156.
Tang M, Shekhar R, Miranda D, Huang D . Characteristics of keratoconus and pellucid marginal degenration in mean curvature maps. Am J Ophthalmol 2005; 140 (6): 993–1001.
Meghpara B, Nakamura H, Vemuganti GK, Murthy SI, Sugar J, Yue BY et al. Histopathologic and immunuhistochemical studies of keratoglobus. Arch Ophthalmol 2009; 127 (8): 1029–1035.
Wong TTL, Sethi C, Daniels JT, Limb GA, Murphy G, Khaw PT . Matrix metalloproteinases in disease and repair processes in the anterior segment. Surv Ophthalmol 2002; 47 (3): 239–256.
Krachmer JH, Feder RS, Belin MW . Keratoconus and the related non-inflammatory corneal thinning disorders. Surv Ophthalmol 1984; 28 (4): 293–322.
Pullum KW, Buckley RJ . A study of 530 patients referred for rigid gas permeable sclera contact lens assessment. Cornea 1997; 16 (6): 612–622.
Foss AJ, Trodd TC, Dart JK . Current indication for sclera contact lenses. CLAO J 1994; 20 (2): 115–118.
Jacobs DS . Update on sclera lenses. Curr Opin Ophthalmol 2008; 19 (4): 298–301.
Bromley JG, Randleman JB . Treatment strategies for corneal ectasias. Curr Opin Ophthalmol 2010; 21 (4): 255–258.
O′donnell C, Welham L, Doyle S . Contact lens management of keratectasia after laser in situ keratomileusis for myopia. Eye Contact Lens 2004; 30 (3): 144–146.
Katsoulos C, Nick V, Lefteris K, Theodore M . Fitting the post-keratoplasty cornea with hydrogel lenses. Cont Lens Anterior Eye 2009; 32 (1): 22–26.
Panda A, Aggarwal A, Madhavi P, Wagh VB, Dada T, Kumar A et al. Management of acute corneal hydrops secondary to keratoconus with intracameral injection of sulfur hexafluoride (SF6). Cornea 2007; 26 (9): 1067–1069.
Sii F, Lee GA, Gole GA . Perforated corneal hydrops treated with sulfur hexafluoride (SF6) gas and tissue adhesive. Cornea 2005; 24 (4): 503–504.
Shah SG, Sridhar MS, Sangwan VS . Acute corneal hydrops treated by intracameral injection of perfluoropropane (C3F8) gas. Am J Ophthalmol 2005; 139 (2): 368–370.
Kaushal S, Sharma N, Vajpayee RB . Treament of acute corneal hydrops with intracameral C3F8 in a patient with pellucid marginal degeneration with keratoglobus. Clin Exp Ophthalmol 2007; 35 (8): 697–699.
Miyata K, Tsuji H, Tanabe T, Mimura Y, Amano S, Oshika T . Intracameral air injection for acute hydrops in keratoconus. Am J Ophthalmol 2002; 133 (6): 750–752.
Basu S, Vaddavalli PK, Ramappa M, Shah S, Murthy SI, Sangwan VS . Intracameral perfluoropropane gas in the treatment of acute corneal hydrops. Ophthalmology 2011; 118 (5): 934–939.
Hussin HM, Biswas S, Majid M, Haynes R, Tole D . A novel technique to treat traumatic corneal perforation in a case of presumed brittle cornea syndrome. Br J Ophthalmol 2007; 91 (3): 399.
Khodadoust AA, Silverstein AM . Studies on the nature of the privilege enjoyed by corneal allografts. Invest Ophthalmol 1972; 11 (3): 137–148.
Cowden JW, Copeland RA, Schneider MS . Large diameter therapeutic penetrating keratoplasties. Refract Corneal Surg 1989; 5 (4): 244–248.
Kodjikian L, Baillif S, Burillon C, Grange JD, Garweg JG . Keratoglobus surgery: penetrating keratoplasty redux. Acta Ophthalmol Scand 2004; 82 (5): 625–627.
Kaufman HE, Werblin TP . Epikeratophakia for the treatment of keratoconus. Am J Ophthalmol 1982; 93 (3): 342–347.
Morgan KS, Stephenson GS, McDonald MB, Kaufman HE . Epikeratophakia in children. Ophthalmology 1984; 91 (7): 780–784.
Morgan KS, McDonald MB, Hiles DA, Aquavella JV, Durrie DS, Hunkeler JD et al. The nationwide study of epikeratophakia for aphakia in older children. Ophthalmology 1988; 95 (4): 526–532.
Javadi MA, Kanavi MR, Ahmadi M, Yazdani S . Outcomes of epikeratoplasty for advanced keratoglobus. Cornea 2007; 26 (2): 154–157.
Morgan KS, Beuerman RW . Interface opacities in epikeratophakia. Arch Ophthalmol 1986; 104 (10): 1505–1508.
Macsai MS, Lamley HL, Schwartz T . Management of oculus fragilis in Ehlers-Danlos type VI. Cornea 2000; 19 (1): 104–107.
Jones DH, Kirkness CM . A new surgical technique for keratoglobus - tectonic lamellar keratoplasty followed by secondary penetrating keratoplasty. Cornea 2001; 20 (8): 885–887.
Burk RO, Joussen AM . Corneoscleroplasty with maintenance of the angle in two cases of extensive corneoscleral disease. Eye 2000; 14 (2): 196–200.
Vajpayee RB, Bhartiya P, Sharma N . Central lamellar keratoplasty with peripheral intralamellar tuck: a new surgical technique for keratoglobus. Cornea 2002; 21 (7): 657–660.
Kaushal S, Jhanji V, Sharma N, Tandon R, Titiyal JS, Vajpayee RB . “Tuck-In” lamellar keratoplasty (TILK) for corneal ectasias involving corneal periphery. Br J Ophthalmol 2008; 92 (2): 286–290.
Kanellopoulos AJ, Pe LH . An alternative surgical procedure for the management of keratoglobus. Cornea 2005; 24 (8): 1024–1026.
Riss S, Heindl LM, Bachmann BO, Kruse FE, Curseifen C . Pentacam-based big bubble deep anterior lamellar keratoplasty in patients with keratoconus. Cornea 2012; 31 (6): 627–632.
Gharbiya M, Moramarco A, Castori M, Parisi F, Celletti C, Marenco M et al. Ocular features in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: a clinical and in vivo confocal microscopy study. Am J Ophthalmol 2012; 154 (3): 593–600.
Nemet AY, Assia EL, Appla DJ, Baraquet AS . Current concepts of ocular manifestations in Marfan’s syndrome. Surv Ophthalmol 2006; 51 (6): 561–575.
van Genderen MM, Kinds GF, Riemslag FC, Hennekam RC . Ocular features in Rubenstein-Taybi syndrome: investigation of 24 patients and review of the literature. Br J Ophthalmol 2000; 84 (10): 1177–1184.
Hennekam RC . Rubenstein-Taybi syndrome. Eur J Hum Genet 2006; 14 (9): 981–985.
van Dijk FS, Cobben JM, Kariminejad A, Maugeri A, Nikkels PG, van Rijn RR et al. Osteogenesis imperfecta: a review with clinical examples. Mol Syndromol 2011; 2 (1): 1–20.
Kiss S, Damico FM, Young LH . Ocular manifestations and treatment of syphilis. Semin Ophthalmol 2005; 20 (3): 161–167.
Aldave AJ, King JA, Cunningham ET Jr. . Ocular syphilis. Curr Opin Ophthalmol 2001; 12 (6): 433–441.
Margo CE, Hamed LM . Ocular syphilis. Survey Ophthalmol 1992; 37 (3): 203–220.
Verity DH, Rose GE . Acute thyroid eye disease (TED): principles of medical and surgical management. Eye 2013; 27 (3): 308–319.
Mizen TR . Thyroid eye disease. Semin Ophthalmol 2003; 18 (4): 243–247.
Bonini S, Coassin M, Aronni S, Lambiase A . Vernal keratoconjunctivitis. Eye 2004; 18 (4): 345–351.
Kumar S . Vernal keratoconjunctivitis: a major review. Acta Ophthalmol 2009; 87 (2): 133–147.
De Smedt S, Wildner G, Kestelyn P . Vernal keratoconjunctivitis: an update. Br J Ophthalmol 2013; 97 (1): 9–14.
Acknowledgements
We acknowledge the support provided by the Hyderabad Eye Research Foundation, Hyderabad, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Wallang, B., Das, S. Keratoglobus. Eye 27, 1004–1012 (2013). https://doi.org/10.1038/eye.2013.130
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2013.130
Keywords
This article is cited by
-
Multimodal diagnostics for keratoconus and ectatic corneal diseases: a paradigm shift
Eye and Vision (2023)
-
Perforierende Excimerlaser-Keratoplastik nach akutem Keratoglobus im Rahmen einer Osteogenesis imperfecta
Die Ophthalmologie (2023)
-
Treating refractory corneal hydrops in a male patient with vernal keratoconjunctivitis and mental retardation: a case report
BMC Ophthalmology (2022)
