
Abstract
Hirschsprung disease (HSCR) is a major cause of chronic constipation in children. HSCR can be caused by germline mutations in RET and EDNRB. Defining causality of the mutations identified is difficult and almost exclusively based on in silico predictions. Therefore, the reported frequency of pathogenic mutations might be overestimated. We combined mutation analysis with functional assays to determine the frequencies of proven pathogenic RET and EDNRB mutations in HSCR. We sequenced RET and EDNRB in 57 HSCR patients. The identified RET-coding variants were introduced into RET constructs and these were transfected into HEK293 cells to determine RET phosphorylation and activation via ERK. An exon trap experiment was performed to check a possible splice-site mutation. We identified eight rare RET-coding variants, one possible splice-site variant, but no rare EDNRB variants. Western blotting showed that three coding variants p.(Pr270Leu), p.(Ala756Val) and p.(Tyr1062Cys) resulted in lower activation of RET. Moreover, only two RET variants (p.(Ala756Val) and p.(Tyr1062Cys)) resulted in reduced ERK activation. Splice-site assays on c.1880-11A>G could not confirm its pathogenicity. Our data suggest that indeed almost half of the identified rare variants are proven pathogenic and that, hence, functional studies are essential for proper genetic counseling.
Similar content being viewed by others


Introduction
Hirschsprung’s disease (HSCR) is a congenital intestinal malformation, characterized by the absence of enteric ganglia in the submucosal and myenteric plexus of the gastrointestinal tract. The incidence of HSCR is estimated at 1 per 5000 live birth. However, it varies significantly between ethnic groups. The highest incidence is reported in the Asian population, 2.8 per 10 000 live birth, while in other ethnic groups, such as Caucasian-Americans, African-Americans and Hispanics, the frequencies are 1.0, 1.5 and 2.1 per 10 000 live birth, respectively.1, 2 In previous studies, several genes have been identified to be responsible for HSCR.2 RET is considered to be the major susceptibility gene for HSCR.2, 3, 4, 5 Extensive studies on RET identified hundreds of coding sequence (CDS) mutations in patients with HSCR. Heterozygous RET CDS mutations have been reported in 7–35% of sporadic cases and in up to 50% in familial cases.6, 7 A wide variety of mutations in RET is found and they are scattered throughout the entire CDS.2 The majority of identified mutations can be divided into two groups, those that result in an early truncation of the protein such as nonsense and frameshift mutations and those that result in a non-functional or less functional protein, which is the case for most missense mutations and small in-frame deletions.8 In general, it is assumed that all mutations found in HSCR patients result in a loss of function.8 However, gain-of-function mutations have also been found in HSCR cases. The majority of patients with gain-of-function mutations suffered, besides HSCR, also from multiple endocrine neoplasia type 2 A (MEN2A) or familial medullary thyroid carcinoma (MTC).9 Not many studies, however, determined the functional consequences these mutations really have on the encoded RET protein.10, 11, 12 Most reports stated their classification of pathogenicity only on in silico predictions. Of the 49 RET variants that have been functionally tested, 32 (65%) were considered disease-causing variants. The rest of those variants (35%) were believed to be non-causative variants.13, 14, 15, 16 It can therefore not be excluded that part of the reported mutations merely are benign, rare polymorphic variants.
The Endothelin B Receptor (EDNRB) is the second most mutated gene after RET in HSCR. Heterozygous EDNRB mutations can predispose to isolated HSCR with incomplete penetrance, while homozygous EDNRB mutations (or compound heterozygous mutations) can result in a more complex phenotype, comprising of HSCR and features of Shah-Waardenburg.17 It is reported that mutations in the EDNRB gene are found in 5–7% percent of HSCR patients and the length of the aganglionosis is variable.2, 18 Functional studies on EDNRB mutations have hardly been conducted. Therefore, comparable with RET, it is unclear whether indeed all reported EDNRB mutations are truly pathogenic. This made us hypothesize that the reported frequency of pathogenic RET and EDNRB mutations might well be overestimated. To prove our hypothesis, we performed an unbiased study in which we combined mutation analysis by regular Sanger sequencing with different kinds of functional assays to determine the real frequency of pathogenic mutations in the Javanese HSCR patient population from Indonesia.
Materials and methods
Patients, parents and controls
We included 57 non-syndromic Javanese HSCR patients and parents of 41 patients from Central Java (Indonesia). The diagnosis HSCR was based on physical examination, radiology findings and histological examination of colon biopsies. Ten milliliter blood were collected from each individual from which we subsequently isolated DNA. Written informed consent was obtained from parents before tissue and blood sampling. Sixty-two unrelated Javanese healthy controls from the Central Java province (Indonesia) were included as controls. This study was approved by the Ethic Committee of the Faculty of Medicine, Universitas Gadjah Mada.
DNA isolation
Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). For DNA isolation out of fresh gut tissues, we used the QIAamp DNA Mini Kit (Qiagen, Düsseldorf, Germany) according to the manufacturer’s protocol.
Mutation analysis
Primers used to amplify all exons of RET (ENST00000355710) and EDNRB (ENST00000334286) are listed in Supplementary Tables S1–S2. All exons were amplified using a touch-down PCR method with annealing temperature ranging from 70 to 60 °C. Sequencing was performed (forward and reverse) with Big Dye Terminator v3.1 Kit (Applied Biosystems, Foster City, CA, USA) on an ABI 3130XL automated sequencer.
In silico prediction
In silico analysis, to predict the functional impact of the mutations identified, was performed using three online software programs: Mutation Taster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/). Algorithmic model used by each software was described by Dong et al.19
Site-directed mutagenesis
All mutations were introduced in a pCMV plasmid containing wild type of the RET9 isoform (pCMV-RET-WT) using a site-directed mutagenesis method (Stratagene, Amsterdam Zuidoost, The Netherlands). Primers used to make each mutant are shown in Supplementary Table S3. The RET mutants sequences were checked by Sanger sequencing.
Transfection and GDNF-GFRα treatment
Eight hundred Human Embryonic Kidney (HEK293) cells were plated in six-well plates cultured in Dulbecco’s Modified Eagle’s Medium (Invitrogen, Carlsbad, CA, USA) with 10% of fetal bovine serum (Invitrogen) and 1% of Penicillin/Streptomycin (Invitrogen) for 24 h at 37 °C, 5% CO2. Cells were transfected with 1 μg each of pCMV-RET-WT or mutant, pGNE-GFP and pCMV-GFRα1 construct using 12 μl of GeneJuice transfection reagent (EMD Millipore, Darmstadt, Germany). Two previously reported RET HSCR variants (p.(Leu56Met) and p.(Tyr1062Phe)) were included in this experiment as the non-pathogenic and pathogenic variant control, respectively.13 After 24 h, transfected cells were treated with 50 ng/μl of GDNF (Peprotech EC, London, UK) for 15 min. Untreated transfected cells in another well were used as a control. Cells were lysed with lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 50 mM NaF, 1% TritonX-100 and protease inhibitor cocktail) and cell lysate was collected after centrifugation at 10 000 r.p.m., at 4 °C for 10 min.
Western blotting and densitometry
Thirty microgram of cells lysate were run on a Criterion TGX 4–12% PAGE precast gel (Bio-Rad, Hercules, CA, USA) at 100–120 volt for 1.5 h. Proteins were transferred onto Nitrocellulose membranes using a semi-dry TransBlot (Bio-Rad) system, using 15 volt for 10 min. Membranes were blocked with 5% skim milk in washing buffer (1 × PBS and 0.1% of Tween-20) at room temperature for 1 h. Membranes were incubated with primary antibodies anti-RET, -ERK, -phosphorylated-RET (p-RET), -phosphorylated-ERK (p-ERK), -GFP and -β-actin in blocking buffer for 24 h at 4 °C. The membranes were washed and incubated with secondary antibodies IRDDye Goat anti-Rabbit and 800 CW Goat anti-Mouse in blocking buffer for 1 h (see list of antibodies on Supplementary Table S4). Membranes were visualized and the densitometry of each band was measured using an Odyssey CLx Infrared Imaging System (Li-Cor, Homburg, Germany). The density of the RET, p-RET and p-ERK bands for all constructs were normalized by the density of the corresponding GFP and β-actin signals. The density of normalized p-RET and p-ERK were further normalized against the normalized RET values. The average of the presented values resulted from three independent experiments are shown in Figure 1b and c.

(a) Western blot analysis of cell lysates isolated from transfected HEK293 cells with pCMV-RET-WT or pCMV-RET-mutants: p.(Arg79Trp), p.(Arg144His), p.(Pro270Leu), p.(Arg694Gln), p.(Ala756Val), p.(Gly533Ser), p.(Tyr1062Cys), p.(Asp489Asn). RET variants p.(Leu56Met) and p.(Tyr1062Phe) were included as controls. (b) Densitometry analysis of p-RET and RET. Values were normalized using GFP and β-actin. (c) Densitometry analysis of p-ERK and RET. Values were normalized using GFP and β-actin. The expression of p-RET or p-ERK is showed as a fold change to RET expression. Three independent experiments were performed (N=3). (*P<0.05, **P<0.01, (−): without GDNF treatment, (+): with GDNF treatment).
Statistical analysis
Results of densitometry of western blotting are presented as mean±standard deviations. Differences of the density of p-RET and p-ERK of the RET mutant proteins, compared with RET-WT, were tested using an unpaired student’s T-test. Differences were regarded as statistically significant when the P-value<0.05.
Exon trap assay
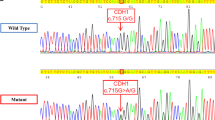
To determine the effect of a possible splice-site variant (c.1880-11 A>G), exon 11 of RET and its flanking region (±150 bp) containing the wild type and the mutant allele were amplified using forward primer (5′GCGA-GAATTC-GGCGGAATTCGGCAGTAAATGGCAGTACCC3′) and reverse primer (5′GCGA-GGATTC-GACGGGATCCACAGACTGTCCCCACACAGC3′). Exon trap assay was performed as previously describe by van der Werft et al.20
Immunofluorescence staining
One hundred HEK293 cells were seeded in a well of a 24-well plate with sterilized and poly-l-lysine-coated coverslips and transiently transfected with 0.4 μg of pCMV-RET-WT or pCMV-RET-p.(Pro270Leu) using 2 μl of GeneJuice transfection reagent according to the manufacturer's instructions. Twenty-four hours after transfection, cells were fixed with 4% PFA for 5 min at room temperature and rinsed three times with 1 × PBS. Cells were blocked with blocking buffer (1% bovine serum albumin and 0.1% Tween-20 in 1 × PBS) for 10 min, washed and incubated overnight at 4 °C with anti-RET antibody (Supplementary Table S4). Cells were washed and incubated for 1 h at room temperature with Cy3 anti-rabbit antibody (1:200, Invitrogen). Cells were washed and incubated for 10 min with Hoechst, rinsed and mounted with ProLong Gold reagent (Thermo Scientific, Waltham, MA, USA). Imaging was done using a Zeiss microscope at × 63 magnification (Zeiss, München, Germany).
Results
Patients' characteristic
Fifty-seven Javanese (Indonesian) patients with HSCR enrolled in this study, 41 male (72%) and 16 female (28%). The age of diagnosis ranges from 1 week to 15 years. Short segment HSCR was seen in 56 cases and 1 patient had a total colonic aganglionosis. Fifty-six cases were sporadic and one case was familial (see Table 1). Of this familial case, we had only DNA of one child available to perform our studies (Patient ID: 45KA).
RET rare coding variants
Nine RET variants with a minor allele frequency (MAF)<0.005 were identified resulting in a variation rate of 15.7% (9/57). The eight CDS-RET variants were all missense variants and one was a possible splice-site variant. None of these variants were present in the Javanese control population. Four missense variants: p.(Arg144His), p.(Ala756Val), p.(Arg79Trp) and p.(Pro270Leu) and the possible splice-site variant, c.1880-11A>G, have never been reported before (novel). The other four missense variations: p.(Asp489Asn), p.(Gly533Ser), p.(Arg694Gln) and p.(Tyr1062Cys) have been reported in the previous studies.11, 12, 14, 21 Seven out of the eight variants were heterozygous, whereas the remainder and the possible splice-site mutation was homozygous. Three out of eight missense variants were de novo variants: p.(Asp489Asn), p.(Ala756Val) and p.(Arg694Gln). The variants found were scattered over the entire gene, and based on location in the gene, we identified that four variants (p.(Arg79Trp), p.(Arg144His), p.(Pro270Leu) and p.(Asp489Asn)) are located in the Cadherin Like-domain, one variant (p.(Gly533Ser)) is located in the Cysteine-rich domain, one variant (p.(Arg694Gln)) is located between the transmembrane domain and the tyrosine kinase (TK) domain, and the last two variants (p.(Ala756Val) and p.(Tyr1062Cys)) are located in the TK domain (Table 2). None of these rare RET variants was identified in our familial case.
RET common variants
Common RET variants were also detected in 56 HSCR patients. Most of those variants are silence variant (c.135G>A, c.1296 G>A, c.2307T>G and c.2712C>G). There were two novel RET variants identified, one of them is a silence variant located in the exon 17 (c.2819T>A) and the other one is a possible splice-site variant located 24 bp upstream of the exon 15 (c.2608-24G>A). Of all those RET common variants identified, only two are missense variants (c.2071G>A/ p.(Gly691Ser) and c.2944C>T/ p.(Arg982Cys)). In the familial case, we identified only a silence RET variant (c.1296G>C) with MAF=0.2248 in 1000 genome database. All RET rare and common variants identified this study are presented in the Supplementary data (Supplementary Table S5). The RET variants identified in this study have been submitted to (http://www.genomed.org/lovd2/submitters_variants.php?submitterid=00041).
EDNRB variants
We identified one variant in the exon 1 (c.143C>T, p.(Pro48Leu) of EDNRB with a MAF<0.005 in the 1000 genome database. However, this variant was also detected in one of our control chromosomes (MAF=0.008) (Table 3). As the MAF in Javanese is >0.005, we decided not to continue with the functional analysis for this variant. None of the rare EDNRB variant was identified in familial case. As for common variant, we identified a previously described silence common variant c.831 A>G located in exon 4 in 27 patients (47.37%). All EDNRB rare and common variants are presented in the Supplementary data (Supplementary Table S6). The EDNRB variants identified in this study have been submitted to (http://grenada.lumc.nl/LOVD2/WS/submitters_variants.php?submitterid=00004).
In silico analysis of coding variants
In silico analysis were performed to predict the pathogenicity of the coding variants. PolyPhen-2 predicted that six out of eight RET missense variants were either possibly or probably damaging (75%), whereas SIFT and Mutation Taster predicted four (50%) and five (62.5%) out of eight RET missense variants were to be damaging (50%). The missense variant identified in EDNRB was predicted to be benign by SIFT and Polyphen-2, however, it was predicted to be disease-causing by Mutation Taster (Tables 2 and 3).
Functional analysis of rare RET-coding variants
GDNF activation
Expression of p-RET and p-ERK for each RET mutant protein (after GDNF stimulation) are presented as fold changes compared with RET-WT expression. The western blot result was shown in Figure 1a. The densitometry data showed that only two RET mutant proteins (p.(Ala756Val) and the positive control p.(Tyr1062Phe) had significantly lower levels of p-RET compared with RET-WT (Figure 1b). While RET-p.(Tyr1062Cys) mutant protein showed a slight difference of p-RET expression (P-value=0.056), all three protein mutants did have significantly lower level of p-ERK (a downstream effector of RET) compare with RET-WT (Figure 1c). On the contrary, p.(Arg79Trp), p.(Arg144His), p.(Pro270Leu), p.(Gly533Ser), p.(Arg694Gln) and the negative control p.(Leu56Met) showed no significant difference in RET or ERK phosphorylation (Figure 1b and c).
Immunofluorescence staining
The extracellular rare variant p.(Pro270Leu) showed a slight reduction of glycosylated/mature RET (170 kDa) protein, when compared with RET-WT or the other mutant counterparts (Figure 1a). The results of the immunofluorescence staining show that the RET-WT protein is localized in the cytoplasm as well as in the cell membrane (Figure 2a). The RET-p.(Pro270Leu) protein localized mostly near the endoplasmic reticulum and the nucleus, rather than in the cytoplasm (Figure 2).

Immunofluorescence staining using a primary antibody against RET upon transfections in HEK293 cells with: (a) pCMV-RET-WT and (b) pCMV-RET-p.(Pro270Leu). RET-WT staining shows strong signals in the cytoplasmic (empty arrow) and cell membrane (arrow heads), whereas the expression of RET-p.(Pro70Leu) is less pronounced and localized closer to the nucleolus/endoplasmic reticulum (arrow).
Exon trap assay
In silico analysis using software (http://www.fruitfly.org/seq_tools/splice.html) suggested that the c.1880-4 A>G variant might reduce the splicing efficiency, however, the exon trap experiment did not show any difference in the splicing pattern between the wild type and the variant (data was not shown).
Discussion
Many groups have searched for disease-causing mutations in a specific gene for a particular disease, resulting in a presumed mutation frequency for that disease in that gene in a (specific) patient population. Almost all these studies presumed pathogenic mutations on in silico predictions. Hardly any of these studies confirmed their finding with functional studies. Whether or not the mutations identified are indeed pathogenic, and the frequencies given are correct, can be debated. In this study, we performed mutation screening on RET and EDNRB in a Javanese patient population, the biggest ethnic group in Indonesia. We further confirmed the level of pathogenicity of the rare variants identified by in vitro studies.
Rare RET variants
In total, nine rare RET variants were identified in 57 HSCR patients (giving a variation rate of 15.8%). This variation rate is more or less similar to reported frequencies in other populations, including Caucasian, Chinese, Korean, Vietnamese and Japanese.2, 10, 12, 22 According to the dysfunctional mechanism and location, the RET missense mutations can be classified into three main groups.23 Those that disturb the maturation (glycosylation) of the protein are located in the Cadherin domain. Mutations that disturb the dimerization of the RET protein upon ligand activation are located in the Cysteine-rich domain. Finally, those that disturb the activity (phosphorylation) of the mutant proteins which are located in the TK domain.23
RET variants in the extracellular part of the protein
Four variants (p.(Arg79Trp), p.(Arg144His), p.(Pro270Leu) and p.(Asp489Asn)) are located in the cadherin domain. Western blot results indicate that only p.(Pro270Leu) showed less mature RET protein (the upper band of 170 kDa) compared with that of RET-WT (Figure 1a). Non-glycosylated protein leads to protein mislocalization owing to incorrect folding.24 We could confirm this by immunofluorescence staining that RET-p.(Pro270Leu) indeed localizes near to the nucleus and the endoplasmic reticulum when compared with RET-WT, which is seen mainly in the cytoplasm and near the cell membrane (Figure 2). Although p-RET and p-ERK expression of RET-p.(Pro270Leu) was not significantly lower compared with RET-WT, this mutant likely leads to protein mislocalization, which will result in reduced or abnormal functioning. The other three cadherin domain variants and the variant in the cysteine-rich domain (p.(Gly533Ser) did not show significantly lower p-RET expression compared with RET-WT upon GDNF stimulation. Variant p.(Asp489Asn), in agreement with the functional study that has been performed by Ishii K et al14 in 2012, indeed did not disturb maturation of the RET protein. However, Ishii et al showed that when combining this variant with two other variants (p.(Val778Asp) and c.2307 T>G)), as observed in a patient, they did see that the mature form of RET protein was significantly decreased.14 How these two intracellular variants in combination with p.(Asp489Asn) could synergistically disturb RET maturation is not known.
RET variants that could result in activated RET
The RET p.(Arg694Gln) de novo missense variant was identified in a 3-week-old male patient with isolated HSCR. This variant was reported previously in a 23-year-old woman with hypothyroidism due to atrophic Hashimoto’s thyroiditis and primary ovarian failure. No family history of familial MTC, MEN2 A and MEN2B, or HSCR was found in that patient.25 Previous functional studies showed that this variant resulted in a gain of function, comparable with what is seen for the MEN2/familial MTC-associated RET mutations.21 Our analysis demonstrated no detectable effects on maturation or phosphorylation of the mutant RET protein. We could therefore not confirm their findings. Activating RET mutations have been found previously in HSCR but were mostly found in the context of MEN2/familial MTC (eg, p.Cys610/618/620).9 For all activating RET mutations, found in HSCR patients, it was shown that the mutant protein besides being activated, also results in a disturbed glycosylation. Our analysis did not show any difference in the glycosylation status. How and whether this de novo rare variant really results in HSCR is therefore unknown. However, one can also not exclude that atrophic Hashimoto’s thyroiditis, primary ovarian failure or HSCR are not correlated to the mutation. Furthermore, we identified a common RET variant p.(Gly691Ser) in this region in six patients. This variant had been reported in previous studies as a modifier for the onset of MEN2A and as a low penetrance variant for MTC. Furthermore, functional studies performed by Colombo et al16 showed that indeed p.(Gly691Ser) has oncogenic potential. Patients who have this variant in combination with proven MEN2A mutation might have an increased risk in getting MEN2A or MTC later on in life.
RET variants in the TK domain
The RET variants p.(Ala756Val) and p.(Tyr1062Cys) are located in the TK domain. Tyrosine 1062 is a known docking site for the Shc adaptor protein. Defect in this particular site disturbs phosphorylation of the tyrosine residue and leads to an inhibition of Shc adaptor protein recruitment.15, 26 As predicted, indeed, p.(Tyr1062Cys) and p.(Ala756Val) disturbed RET phosphorylation and subsequently lead to the inhibition of ERK phosphorylation (Figure 1a and c). How the p.(Ala756Val) variant leads to reduced RET phosphorylation is unknown. This variant is located rather close to the adenosine triphosphate binding site which is conserved in TKs.15 This mutant might disturb the adenosine triphosphate binding to the TK domain of RET which could lead to the failure of phosphorylation of the closest tyrosine/threonine residue.
De novo RET variants
De novo appearance is often considered a strong argument in favor of pathogenicity. But is this assumption correct? Our functional assays confirm pathogenicity for only one (p.(Ala756Val) of the three de novo variants identified. For the other two de novo variants, p.(Asp489Asn) and p.(Arg694Gln), we did not find any evidence for pathogenicity. Are these two de novo variants therefore not pathogenic? From a statistical point of view, one could argue that finding a de novo mutation in a disease-causing gene by chance is extremely low. Assuming that, on average, each individual has a little more than one de novo heterozygous in the coding region and assuming ~21 000 genes in duplicate, finding a de novo RET variant by chance is ~1 in 40 000. Therefore, statistically, it is likely that these variants cause or contribute to disease development. This is further strengthened by the fact that both p.(Asp489Asn) and p.(Arg694Gln) have been found before in the context of HSCR and therefore are recurrent HSCR rare variants. The combination of a de novo appearance and the fact that they are recurrent make it extremely likely that these variants are pathogenic even when no functional effects are found.27
Common RET variants
Several common variants (c.135G>A, c.1296 G>A, c.2307 T>G and c.2712C>G) were identified, for which the frequencies of those polymorphic alleles were similar to those described in other populations.11, 12, 26 Garcia-Barcelo et al28 showed that the haplotype containing three common RET variants (c.135G>A, c.1296G>A and c.2307 T>G) are represented in 66% of HSCR patients and c.1296G> A is associated with a more severe form of HSCR. As all but one of the HSCR patients included in this study have the same phenotype, the less severe form of the disease, namely, short-segment of HSCR, we could not confirm this association in our patient cohort. However, we did observe that all but one of the patients who carry this haplotype (n=11, 19%) do not have a rare RET variant. This might indicate that this haplotype has a strong modifier effect, and hence, combinations of this haplotype with a mutation in a non-RET gene might lead to the development of HSCR.
EDNRB variants
The variation rate of EDNRB in other populations is considered around ~5%.2, 18 Surprisingly, the mutation rate of EDNRB in our Javanese population seems to be much lower than those in Caucasian, Chinese or in any other population.29, 30 This might be because of the small sample size, but it might also indicate that EDNRB is not a major gene for HSCR in Javanese population. Study on a larger cohort of Javenese HSCR patients needs to be performed to confirm this hypothesis.
Conclusion
This study shows that only three out of nine CDS RET variants (p.(Pro270Leu), p.(Ala756Val) and p.(Tyr1062Cys)) showed clear pathogenic effects. One could argue that in addition also the two de novo variants (p.(Asp489Asn) and p.(Arg694Gln)), for which we did not detect any pathogenic effect on RET function, are pathogenic because they are recurrent mutations in HSCR. Even doing so, it leaves us with four CDS missense mutations for which we cannot confirm a pathogenic role. In this study, we only looked for glycosylation and phosphorylation of the RET protein and to downstream signaling of RET via ERK. RET also activates many other downstream targets which were not analyzed in this study. One cannot exclude that signaling via these other pathways is disturbed. Moreover, functional studies for complex diseases such as HSCR are challenging, as the disease, in most cases, is caused by a combination of variants, all with a modest effect. The functional studies we performed are sensitive enough to measure strong effects of a single variant, but likely are not suitable to detect modest to mild effects. Effect as a result of combinations of variants in the same gene can be measured with our assays. This type of functional studies had been performed in previous studies on RET variants both in HSCR and MEN2A and showed a clear synergetic effect of combined mutations.14, 16 On the other hand, one cannot exclude that it is the combination of two variant alleles of the same gene (not in cis) that give the effect or that it is the effect of variants in different genes that give rise to the HSCR phenotype. In silico analysis using online software proved useful, especially when we are dealing with many variants to be functionally studied. In this study, Mutation Taster accurately predicted for the three variants proven to be pathogenic by functional studies as disease-causing variants. However, from our study, it became clear that different software packages gave different results, although the basic principles they use for their logarithms is more or less similar. Hence, when using in silico prediction programs, we advise to use more than one prediction software to minimize a wrong outcome. In summary, if we consider only the pathogenic variants based on our functional studies, the mutation rate of RET in the Javanese patient population is ~5% (3/57). If we include all de novo variants, it is ~9% (5/57). However, almost half of the variants identified cannot be considered pathogenic. This has a clear impact on genetic counseling. Assuming that our findings can be generalized, this indicates that likely the frequencies of clear pathogenic RET mutations in HSCR, as reported previously, are overestimated. Therefore, one should be cautious using RET mutations for genetic counseling, when they are not functionally tested. We recommend that when mutation data are used for important clinical decision making, such as prenatal diagnostics, functional analysis is warranted.
References
Angrist M, Bolk S, Thiel B et al: Mutation analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet 1995; 4: 821–830.
Amiel J, Sproat-Emison E, Garcia-Barcelo M et al: Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 2008; 45: 1–14.
Brooks AS, Oostra BA, Hofstra RM : Studying the genetics of Hirschsprung’s disease: Unraveling an oligogenic disorder. Clin Genet 2005; 67: 6–14.
Garcia-Barceló M, Sham MH, Lee WS et al: Highly recurrent RET mutations and novel mutations in genes of the receptor tyrosine kinase and endothelin receptor B pathways in Chinese patients with sporadic Hirschsprung disease. Clin Chem 2004; 50: 93–100.
Martucciello G, Ceccherini I, Lerone M, Jasonni V : Pathogenesis of Hirschsprung’s disease. J Pediatr Surg 2000; 35: 1017–1025.
Hofstra RM, Wu Y, Stulp RP et al: RET and GDNF gene scanning in Hirschsprung patients using two dual denaturing gel systems. Hum Mutat 2000; 15: 418–429.
Attié T, Pelet A, Edery P et al: Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet 1995; 4: 1381–1386.
Pasini B, Borrello MG, Greco A et al: Loss of function effect of RET mutations causing Hirschsprung disease. Nat Genet 1995; 10: 35–40.
Sijmons RH, Hofstra RM, Wijburg FA et al: Oncological implications of RET gene mutations in Hirschsprung’s disease. Gut 1998; 43: 542–547.
Kim JH, Yoon KO, Kim JK et al: Novel mutations of RET gene in Korean patients with sporadic Hirschsprung’s disease. J Pediatr Surg 2006; 41: 1250–1254.
Sangkhathat S, Kusafuka T, Chengkriwate P, Patrapinyokul S, Sangthong B, Fukuzawa M : Mutations and polymorphisms of Hirschsprung disease candidate genes in Thai patients. J Hum Genet 2006; 51: 1126–1132.
Ngo DN, So MT, Gui H et al: Screening of the RET gene of Vietnamese Hirschsprung patients identifies 2 novel missense mutations. J Pediatr Surg 2012; 47: 1859–1864.
Geneste O, Bidaud C, De Vita G et al: Two distinct mutations of the RET receptor causing Hirschsprung’s disease impair the binding of signalling effectors to a multifunctional docking site. Hum Mol Genet 1999; 8: 1989–1999.
Ishii K, Doi T, Inoue K, Okawada M et al: Correlation between multiple RET mutations and severity of Hirschsprung’s disease. Pediatr Surg Int 2013; 29: 157–163.
Iwashita T, Kurokawa K, Qiao S et al: Functional analysis of RET with Hirschsprung mutations affecting its kinase domain. Gastroenterology 2001; 121: 24–33.
Colombo C, Minna E, Rizzetti MG et al: The modifier role of RET-G691S polymorphism in hereditary medullary thyroid carcinoma: functional characterization and expression/penetrance studies. Orphanet J Rare Dis 2015; 10: 25.
McCallion AS, Chakravarti A : EDNRB/EDN3 and Hirschsprung disease type II. Pigment Cell Res 2001; 14: 161–169.
Kusafuka T, Wang Y, Puri P : Mutation analysis of the RET, the endothelin-B receptor, and the endothelin-3 genes in sporadic cases of Hirschsprung’s disease. J Pediatr Surg 1997; 32: 501–504.
Dong C, Wei P, Jian X, Gibbs R et al: Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2014; 24: 2125–2137.
Van Der Werf CS, Wabbersen TD, Hsiao N et al: CLMP is required for intestinal development, and loss-of-function mutations cause congenital short-bowel syndrome. Gastroenterology 2012; 142: 453–462.
Wu TT, Tsai TW, Chu CT et al: Low RET mutation frequency and polymorphism analysis of the RET and EDNRB genes in patients with Hirschsprung disease in Taiwan. J Hum Genet 2005; 50: 168–174.
Leon TYY, So MT, Lui VCH et al: Functional analyses of RET mutations in Chinese Hirschsprung disease patients. Birth Defects Res A Clin Mol Teratol 2012; 94: 47–51.
Plaza-Menacho I, Burzynski GM, Groot JW, de Eggen BJ, Hofstra RM : Current concepts in RET-related genetics, signaling and therapeutics. Trends Genet 2006; 22: 627–636.
Kjær S, Ibáñez CF : Intrinsic susceptibility to misfolding of a hot-spot for Hirschsprung disease mutations in the ectodomain of RET. Hum Mol Genet 2003; 12: 2133–2144.
Orgiana G, Pinna G, Camedda A et al: A new germline RET mutation apparently devoid of transforming activity serendipitously discovered in a patient with atrophic autoimmune thyroiditis and primary ovarian failure. J Clin Endocrinol Metab 2004; 89: 4810–4816.
Asai N, Murakami H, Iwashita T, Takahashi M : A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with Shc adaptor proteins. J Biol Chem 1996; 271: 17644–17649.
So MT, Leon TY, Cheng G et al: RET mutational spectrum in Hirschsprung disease: Evaluation of 601 Chinese patients. PLoS One 2011; 6: e28986.
Garcia-Barceló MM, Sham M, Lui VC et al: Chinese patients with sporadic Hirschsprung’s disease are predominantly represented by a single RET haplotype. J Med Genet 2003; 40: e122.
Gath R, Goessling A, Keller KM et al: Analysis of the RET, GDNF, EDN3, and EDNRB genes in patients with intestinal neuronal dysplasia and Hirschsprung disease. Gut 2001; 48: 671–675.
Auricchio A, Casari G, Staiano A, Ballabio A : Endothelin-B receptor mutations in patients with isolated Hirschsprung disease from a non-inbred population. Hum Mol Genet 1996; 5: 351–354.
Acknowledgements
We extend our gratitude to all patients and their family who voluntarily enrolled to this study. This study was supported by research grant from ZonMW (TOP-subsidie 40-00812-98-10042) and the Maag Lever Darm stichting (WO09-62).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Widowati, T., Melhem, S., Patria, S. et al. RET and EDNRB mutation screening in patients with Hirschsprung disease: Functional studies and its implications for genetic counseling. Eur J Hum Genet 24, 823–829 (2016). https://doi.org/10.1038/ejhg.2015.214
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.214
This article is cited by
-
High incidence of EDNRB gene mutation in seven southern Chinese familial cases with Hirschsprung’s disease
Pediatric Surgery International (2024)
-
Circular RNA MTCL1 targets SMAD3 by sponging miR-145‐5p for regulation of cell proliferation and migration in Hirschsprung’s disease
Pediatric Surgery International (2023)
-
Outcome of Clinical Genetic Testing in Patients with Features Suggestive for Hereditary Predisposition to PTH-Mediated Hypercalcemia
Hormones and Cancer (2020)
-
Sequence characterization of RET in 117 Chinese Hirschsprung disease families identifies a large burden of de novo and parental mosaic mutations
Orphanet Journal of Rare Diseases (2019)
-
Advances in understanding functional variations in the Hirschsprung disease spectrum (variant Hirschsprung disease)
Pediatric Surgery International (2017)
