
Abstract
So far, the role of mutations in the δ-sarcogylcan (Sgcd) gene in causing autosomal dominant dilated cardiomyopathy (DCM) remains inconclusive. A p.S151A missense mutation in exon 6 of the Sgcd gene was reported to cause severe isolated autosomal dominant DCM without affecting skeletal muscle. This is controversial to our previous findings in a large consanguineous family where this p.S151A mutation showed no relevance for cardiac disease. In this study, the potential of the p.S151A mutation to cause DCM was investigated by using two different approaches: (1) engineering and characterization of heterozygous knock-in (S151A-) mice carrying the p.S151A mutation and (2) evaluation of the potential of adeno-associated virus (AAV) 9-based cardiac-specific transfer of p.S151A-mutated Sgcd cDNA to rescue the cardiac phenotype in Sgcd-deficient (Sgcd-null) mice as it has been demonstrated for intact, wild-type Sgcd cDNA. Heterozygous S151A knock-in mice developed a rather mild phenotype of cardiomyopathy. Increased heart to body weight suggests cardiac enlargement in 1-year-old S151A knock-in mice. However, at this age cardiac function, assessed by echocardiography, is maintained and histopathology completely absent. Myocardial expression of p.S151A cDNA, similar to intact Sgcd cDNA, restores cardiac function, although not being able to prevent myocardial histopathology in Sgcd-null mice completely. Our results suggest that the p.S151A mutation causes a mild, subclinical phenotype of cardiomyopathy, which is prone to be overseen in patients carrying such sequence variants. Furthermore, this study shows the suitability of an AAV-mediated cardiac gene transfer approach to analyze whether a sequence variant is a disease-causing mutation.
Similar content being viewed by others


Introduction
Familial dilated cardiomyopathy (DCM) is a genetically heterogeneous disease caused by mutations in many different genes including those encoding proteins of the dystrophin-associated glycoprotein complex (DGC). Mutations in these genes may cause disease phenotypes that range from DCM to muscular dystrophy.1 DGC members have a central role in maintaining integrity of the cell membrane by linking the extracellular matrix with the cytoskeleton, thus protecting muscle fibers from contraction-induced damage and necrosis.2 The sarcoglycan subcomplex is an integral part of the DGC with specific compositions that vary between different tissues. In cardiac and skeletal muscle, the DGC complex is primarily composed of the four components α-, β-, γ- and δ-sarcoglycan, essentially involved in sarcolemmal integrity by interaction with integrins that mediates cell adhesion to the extracellular matrix.3 A discussed association of dystrobrevin and the syntrophins with nNOS suggests that the sarcoglycan complex is involved in signal transduction.4 Mutations in the α-, β-, γ- and δ-sarcoglycan genes (Sgca, b, g and d) cause a heterogenous group of autosomal recessive limb girdle muscular dystrophies (LGMD2C–F) clinically characterized by progressive muscle weakness. In particular, LGMD2F, caused by mutations in the Sgcd gene, is frequently associated with potentially fatal DCM.5, 6, 7 δ-Sarcoglycan-deficient mice (Sgcd−/−) develop early progressive cardiomyopathy and muscular dystrophy and serve as a useful animal model for LGMD2F.8, 9
In contrast to autosomal recessive LGMD2F, an autosomal dominant mode of inheritance was suggested for patients with isolated familial DCM caused by mutations in the Sgcd gene.10, 11 Tsubata et al10 found a single missense mutation (451T>G, p.S151A) associated with severe DCM in a family with three patients carrying this mutation. Two mutant carriers died of early sudden cardiac death while the third patient required heart transplantation at young age. The potential pathogenicity of the p.S151A variant was underlined by the finding of early DCM with enhanced lethality in transgenic mice overexpressing the mutated allele up to sevenfold.12 It remained uncertain whether the expression level or the mutant protein was responsible for cardiac pathology in these mice.
Moreover, the pathogenic relevance of the p.S151A variant was challenged when this mutation was discovered in a large consanguineous family homozygous for p.A131P suffering from LGMD2F but lacking any signs of cardiac disease in family members carrying the p.S151A mutation.13 Thus, because of the controversy, the pathogenic potential of the p.S151A missense mutation to cause familial cardiomyopathy has to be defined.
In this study, the potential of this mutation to cause DCM was investigated by two different approaches: (1) engineering and characterization of heterozygous (S151A) knock-in mice and (2) evaluating the potential of mutated p.S151A Sgcd using adeno-associated virus (AAV) vectors to rescue the cardiac phenotype in Sgcd-deficient mice as shown for intact, wild-type Sgcd cDNA.14
The knock-in of p.S151A caused a rather mild phenotype of cardiomyopathy in mice. AAV-mediated myocardial expression of the mutated p.S151A cDNA in Sgcd-deficient mice restored cardiac function similar to wild-type Sgcd cDNA, but was unable to prevent myocardial histopathology completely.
Materials and methods
Engineering of the targeting construct
A 7.2-kb DNA fragment of the Sgcd gene containing intron 5, exon 6 and intron 6 sequences was amplified using genomic DNA from a C57BL/6 mouse and the following oligonucleotides: 5′-ATGCTCCAATTGCCGTGGTC-3′ (forward) and 5′-AGTGAAATCCATGGCGACAAAG-3′ (reverse). This fragment was cloned into vector pCR2.1-TOPO (Invitrogen, Darmstadt, Germany) harboring both an ampicillin (ampR) and a kanamycin (kanR) resistance gene for bacterial selection. A fragment of the KanR was deleted by PstI digestion and religation. A neomycin (neoR) resistance cassette enabling selection in both bacteria and mammalian cells was then introduced as follows: the cassette was amplified from vector pL452 (refs 15,16) using two oligonucleotide combinations: 5′-CTCTGCAAAATGGATCCCCTCGAGGGACCTA-3′ (forward) and 5′-CGATCATATTCAATAACCCTTAAT-3′ (reverse) and 5′-GATCCCCTCGAGGGACCTA-3′ (forward) and 5′-CTGATGGAATGCCGATCATATTCAATAACCCTTAAT-3′ (reverse). The two amplification products were mixed at a 1:1 ratio and denatured and renatured to create heteroduplices with 5′-overhangs. In the same way, recombinant vector pCR2.1-TOPO containing the mouse DNA was amplified using the oligonucleotides 5′-GCATTCCATCAGTGGCGGCATGCCCCAGCTAC-3′ (forward) and 5′-GATACTGAAGCAAGTGCCTGGAAC-3′ (reverse) and 5′-TGGCGGCATGCCCCAGCTAC-3′ (forward) and 5′-CATTTTGCAGAGGATACTGAAGCAAGTGCCTGGAAC-3′ (reverse), respectively, to produce heteroduplices with 5′-overhangs. Owing to the complementary 5′-overhangs (12 bp) of the different heteroduplices, the neoR cassette could be cloned into intron 6 of the pCR2.1-TOPO derivative. Finally, the p.S151A mutation was introduced into exon 6 by site-directed mutagenesis (Quick change XL Site-Directed Mutagenesis kit, Stratagene, La Jolla, CA, USA).
Generation of heterozygous S151A knock-in mice
The targeting vector was linearized and electroporated into murine ES cells from 129/sv mice. G418-FIAU double resistant colonies were picked and screened by PCR amplification and Southern blot hybridization for the targeted p.S151A allele. A positive ES cell clone was injected into blastocysts derived from C57BL/6 mice. A line of mice carrying the mutated p.S151A allele in the germline was established. The chimeras were bred into C57BL/6 mice.
Genotyping of S151A knock-in mice
For identification of heterozygous mice, a multiplex PCR was established with the following oligonucleotides for the wild-type allele (forward 5′-CAGTGAAACGGACCAGTGTG-3′, reverse 5′-CTGGGAAGAAGGCACTTCAG-3′) and for the mutated allele (forward 5′-GGACAGGTCGGTCTTGACA-3′, reverse 5′-CATTCTGCACGCTTCAAAAG-3′). Genotyping results were confirmed by Southern blot analyses. DNA sequence analysis of the entire exon 6 carrying the p.S151A mutation was performed using the following oligonucleotides: forward 5′-TTGTAGTGACGGGCGACAAGG-3′, reverse 5′-GCCTACAAAAGATGGCACCACG-3′. Animals were bred to a C57BL7/6 background for at least six generations and only male mice have been used for the experiments.
Animal procedures
All procedures involving the use and care of animals were performed according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and the German animal protection code. Approval was also granted by the local ethics review board.
Sgcd-deficient mice (B6.129-Sgcdtm1Mcn/J) were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and bred in our animal facility. The virus solution was intravenously injected into the tail vein of 2-month-old mice as 150 μl bolus using a sterile syringe and 29-gauge needle.
RNA isolation and cDNA synthesis
RNA was extracted from different frozen tissue samples applying the TRizol procedure (Invitrogen, Karlsruhe, Germany). Isolated RNAs were then treated with RNAse-free DNAse I to remove residual genomic DNA and cDNA was synthesized using DNAse I-treated RNA, the Superscript III kit (Invitrogen) and random hexamer priming.
Real-time polymerase chain reaction
Expression of brain natriuretic peptide (BNP) in cardiac tissue was quantified at the mRNA level using real-time PCR as previously described.14 For real-time PCR, quantification of GAPDH was used as housekeeping gene. The PCR was run in an ABI PrismTM 7000 Sequence Detection System (Applied Biosystems, Weiterstadt, Germany), and a 7000 System SDS Software was used for data analysis.
Quantitative PCR
For quantification and discrimination of Sgcd wild-type and mutant transcripts (Sgcd-S151A) in heart samples a ABI PrismTM 7000 Sequence Detection System and a custom-made TaqMan assay (Applied Biosystems) containing the oligonucleotides 5′-AAGCATATGGCAAAAGGTTTGAAGTAAAA-3′ (forward) and 5′-CCCACGACCACTTCACTG T-3′ (reverse) and a specific probe for both wild-type (5′-ATCCGCAGAAAAGA-3′) and mutant allele (5′-ATCCGCAGCAAAGA-3′) were utilized (bold letters indicate the site of mutation). Reactions were performed with the Platinum qPCR SuperMix-UDG (Invitrogen) and quantified as previously described.17 The specificity of the TaqMan probes was tested with Sgcd wild-type vector DNA and the targeting vector containing the p.S151A mutation mixed in a ratio 1:1.
Immunohistochemistry, Evans blue dye (EBD) and histological analyses
Hematoxylin and eosin (H&E) and immunofluorescence staining for α-, β-, γ- and δ-sarcoglycan were performed as previously described.14 Briefly, after blocking unfixed 7 μm cryosections were incubated overnight with primary antibodies against Sgca, Sgcb, Sgcg (mouse monoclonal, dilution 1:50, Novacastra, Newcastle, UK) and Sgcd (polyclonal rabbit H55, 1:50, Santa Cruz, Heidelberg, Germany). After incubation with biotinylated anti-mouse or anti-rabbit secondary antibodies (1:100), samples were overlaid with fluorescein–streptavidin (1:100).
In all, 25 μl per 10 g body weight of EBD solution (concentration 0.5 mg EBD/0.05 ml PBS; Sigma Chemical Co., St Louis, MO, USA) were injected intraperitoneally. After 16 h, hearts were extracted and Evans blue-positive fibers were identified by fluorescence microscopy.
Transthoracic echocardiography
Echocardiography was performed using a Sonos 5500 with a S12 transducer (Hewlett-Packard, Palo Alto, CA, USA) (12 MHz). The echocardiographer was blinded with respect to the treatment group. Mice were shaved and left ventricular parasternal short-axis views were obtained in M-mode imaging at the papillary muscle level. Three consecutive beats were used for measurements of left ventricular end diastolic internal diameter (LVEDD) and left ventricular end-systolic internal diameter (LVESD). Fractional shortening (FS) was calculated as FS%=((LVEDD–LVESD)/LVEDD) × 100.
Voluntary wheel running
At the beginning of the study, all treated and untreated wild type, p.S151A knock-in and Sgcd-deficient mice were placed individually in cages containing running wheels (d=11 cm; Fahrholz, Hamburg, Germany). All mice were maintained under a standard 12:12 h light–dark cycle and had free access to the wheel. The individual daily running distance was measured with digital magnetic counters (BC 500, Sigma Elektro GmbH, Neustadt, Germany) connected to the running wheels.
Production of AAV vectors
AAV vector genome plasmids were generated by replacing EGFP of pdsCMV-MLC260-EGFP18 with the mutated p.S151A or wild-type murine δ-sarcoglycan cDNA using BsrGI/NheI restriction sites. pdsCMV.MLC260-EGFP was used for generation of control vector expressing EGFP.
For production of AAV9 pseudotyped vectors, each of these vector genome plasmids was co-transfected with p5E18-VD2-9, containing the AAV9 capsid sequence and the helper plasmid pDGdelVP, resulting in AAV9-Sgcd, AAV9-S151A and AAV9-EGFP. Vectors were purified by a filtration cascade followed by an iodixanol step gradient centrifugation and titrated as described before.19 Transduction efficiency of systemic gene transfer of AAV9-Sgcd or AAV9-S151A was defined as percentage of transduced cardiomyocytes expressing Sgcd localized at the sarcolemma and was assessed semiquantitatively using fluorescence microscopy.
Statistical analysis
All data are reported as mean+(SD). Differences between the animal groups were compared by an unpaired Student’s t-test or using two-factor ANOVA with Scheffe’s test for individual subgroup comparisons. P<0.05 was used as criteria for statistical significance.
Results
Generation of heterozygous S151A knock-in mice
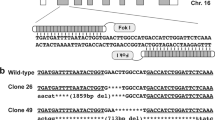
Human δ-sarcoglycan is a type II transmembrane glycoprotein that is mainly expressed in cardiac and skeletal muscle and shares about 90% protein sequence identity with the murine δ-sarcoglycan.20 The serine at codon 151 that was previously found to be changed to an alanine in a DCM family is conserved in both species.10 Therefore, we designed a targeting vector encoding the S151A substitution in exon 6 of the Sgcd gene (451T>G; p.S151A) introducing a neomycin resistance cassette into the intron 6 sequence for positive selection of ES cells as illustrated in Figure 1a. Homologous recombination was confirmed by PCR analyses over the 5′- and 3′- homologous arms, with one oligonucleotide in each reaction matching sequence in the neomycin resistance cassette and the other matching sequence outside the targeting vector. The correct integration was confirmed with Southern blot hybridization using two different radioactive labelled probes, which have been amplified containing intron 6 and neomycin cassette sequences (data not shown). Positive ES cell clones were then injected into blastocysts to obtain germline-competent chimeric mice. A mouse line with stable transmission of the p.S151A mutation was used for subsequent generations (Figure 1b). For breeding, genotypes were determined by PCR amplification from tail DNA. Crossing of heterozygous mice produced no homozygous offspring, suggesting that homozygous embryos died during embryogenesis. Allele-specific quantification of expression of the mutant S151A allele compared with the wild-type allele using a TaqMan assay revealed 4.2±3.1% in hearts and 3.3±3.0% in skeletal muscle of adult heterozygous mice (n=12).

Generation of δ-sarcoglycan targeted S151A knock-in mice. (a) Upper part shows a schematic representation of the targeting vector containing intron 5, exon 6 and intron 6 of the Sgcd gene in addition to a neoR cassette located in intron 6. Middle part shows schematically the homologous recombination event with genomic DNA represented by exon 5, intron 5, exon 6, intron 6 and exon 7 of Sgcd. Lower part shows the recombined S151A allele. Exons 5–7 are depicted by filled boxes. The exon 6 451T>G designates the 451T>G (p.S151A) missense mutation. (b) Sequence analyses in heterozygous S151A knock-in mice revealed compared with wild-type mice a change from thymine to guanine in position 451 in exon 6 of the Sgcd gene (451T>G) leading to an amino acid change at position 151 from serine to alanine (p.S151A).
Inconspicuous cytological phenotype of heterozygous S151A knock-in mice
The sarcoglycan complex is found at the plasma membrane. In 1-year-old heterozygous S151A knock-in mice and age-matched wild-type mice, we found no differences in the expression of α-, β-, γ- and δ-sarcoglycan whereas sarcoglycan expression was absent in Sgcd-deficient mice as expected (Figure 2a).

The sarcolemmal membrane of 1–year-old heterozygous S151A knock-in mice is intact and cardiomyocytes show no morphological changes. (a) Immunofluorescence staining of cardiac sections revealed that α-, β-, γ- and δ-sarcoglycan is present in wild-type and heterozygous S151A knock-in mice, but lacking in homozygous Sgcd-deficient mice. (b) Evans blue staining (red) revealed the absence of EBD-positive fibers in S151A knock-in mice, suggesting preserved integrity of the plasma membrane. In contrast, Evans blue-stained fibers were present in cardiomyocytes of Sgcd-deficient mice. (c) H&E-stained cardiac sections of S151A knock-in and wild-type mice showed no histological signs for DCM such as cell degeneration, necrosis and fibrosis, which clearly were seen in Sgcd-deficient mice. Scale bar: 50 μm.
Weakening of the sarcoglycan complex because of loss-of-function mutations can result in instability of the plasma membrane and increased membrane permeability, which can be visualized by EBD uptake exclusively in the damaged fibers. We analyzed the hearts of 1-year-old heterozygous S151A knock-in and age-matched wild-type mice and found no EBD-positive fibers, suggesting that integrity of the plasma membrane is preserved in the S151A mutant mice. In contrast, the hearts of adult Sgcd-deficient mice showed extensive areas of EBD uptake indicating an increased sarcolemmal damage in cardiomyocytes (Figure 2b).
H&E staining of cardiac sections of 1-year-old heterozygous S151A knock-in mice and age-matched control mice showed no histological signs for DCM such as cell degeneration, necrosis and fibrosis as seen in the Sgcd-deficient mice used as disease model for sarcoglycan cardiomyopathy (Figure 2c).
Heterozygous S151A knock-in mice show a mild phenotype of cardiomyopathy
To verify whether a possible heart or skeletal muscle phenotype could be triggered by physical stress, we exercised 10-month-old S151A knock-in mice and wild-type littermates, respectively, for 22 weeks using a voluntary wheel running system (n=8). From the age of 52 weeks, heterozygous S151A knock-in mice showed a significantly reduced cumulative running distance over a period of 4 weeks compared with age-matched wild-type mice (Supplementary Figure 1). Exercised heterozygous mutant (S151A) and age-matched wild-type mice showed similar body weight but a significantly increased heart to body weight ratio in mutant mice might point to a discrete cardiac enlargement or hypertrophy (Figure 3a). To assess left ventricular function, transthoracic echocardiography was performed in 1-year-old wild type, heterozygous S151A knock-in and Sgcd-deficient mice and revealed no difference in fractional shortening of S151A knock-in and wild-type mice whereas heart function was significantly reduced in Sgcd-deficient mice (Figure 3b). Moreover, Sgcd-deficient mice with their significantly increased BNP mRNA levels,14 clearly differ from S151A mutant mice with BNP mRNA levels similar to wild-type mice (Figure 3c) indicating a preserved left ventricular function in S151A mutant mice. As expression of the mutant allele was low, we additionally phenotyped heterozygous Sgcd+/− mice. As shown in Supplementary Figure 2, Sgcd−/− mice develop signs of mild cardiomyopathy with patchy areas of fibrosis and necrosis in histology (A) and reduced fractional shortening in echocardiography (B), whereas heterozygous Sgcd+/− mice show no signs of cardiomyopathy.

One-year-old heterozygous S151A knock-in mice develop a mild DCM phenotype after voluntary wheel running. (a) Although the body weight was similar to wild-type mice (n=14), the heart to body weight ratio (HW/BW) in heterozygous S151 knock-in (n=11) and Sgcd-deficient mice (n=12) was increased indicating cardiac enlargement (**P<0.001 and ++P<0.001). (b) Based on echocardiographic measurements, there is no significant difference in fractional shortening (FS) between S151A knock-in mice (n=8) and wild-type mice (n=8), but a significant decrease in Sgcd-deficient mice (n=8) (+P<0.01). (c) No significant difference was found between BNP-expression in wild-type (n=6) and S151A knock-in mice (n=6) while Sgcd-deficient mice (n=9) revealed significantly higher levels (++P<0.001; NS, statistically not significant).
AAV9-mediated expression of Sgcd S151A restores ventricular dysfunction but not histopathology in Sgcd-deficient mice
Cardiac gene transfer of Sgcd cDNA with intravenous injection of AAV9 vectors has been found to prevent cardiomyopathy in Sgcd-deficient mice.14 To further investigate the role of the Sgcd S151A variant in DCM we compared it with the wild-type sequence regarding its ability to prevent cardiomyopathy in an AAV9-mediated gene transfer approach. Eight-week-old Sgcd-deficient mice were injected intravenously either with AAV9-S151A or AAV9-Sgcd. After 1 year, immunohistochemistry showed efficient myocardial expression of mutated or wild-type δ-sarcoglycan at the sarcolemma in >80% of cardiomyocytes (Figures 4a and b) and a reconstitution of the sarcoglycan complex reflected by a similar staining with an α-sarcoglycan antibody. Moreover, gene delivery of both AAV9-Sgcd and AAV9-S151A is able to prevent the decline in left ventricular function compared with control-treated mice (Figure 4c). Fractional shortening in AAV9-Sgcd and AAV9-S151A treated Sgcd-deficient mice did not differ from those of wild-type mice, whereas myocardial contractility in Sgcd-deficient mice treated with AAV9-EGFP significantly decreased at the age of 1 year. Histopathological examinations of cardiac sections of control-treated Sgcd-deficient mice showed extensive signs of cardiomyopathy such as fibrosis, cell degeneration and necrosis (Figure 4a). Although there was a distinct amelioration of cardiac pathology in Sgcd-deficient mice treated with AAV9-S151A, cardiac damage could not be prevented completely compared with AAV9-Sgcd-treated animals despite sarcolemmal localization of mutant δ-sarcoglycan (Figure 4a).

AAV9-mediated cardiac expression of the δ-sarcoglycan S151A variant in Sgcd-deficient mice left ventricular function and ameliorates histopathology: (a) H&E staining and immunohistochemistry. Hearts of AAV9-GFP treated Sgcd-deficient mice (control) showed extensive fibrosis, cell degeneration and necrosis, which was absent from AAV9-S151A. A distinct amelioration of cardiac pathology observed in AAV9-S151A and AAV9-Sgcd treated Sgcd-deficient mice yield a pattern comparable to wild-type mice (Scale bar: 50 μm). (b) After systemic cardiac gene transfer >80% of the cardiomyocytes express δ-sarcoglycan in AAV9-S151A (82.8%, n=4) and AAV9-Sgcd (84.0%, n=4) treated Sgcd-deficient mice (NS, statistically not significant). (c) Echocardiographic measurements. Fractional shortening (FS) in 1-year-old AAV9-S151A-treated mice (n=8) is not different from AAV9-Sgcd-treated mice (n=8) or wild-type mice (n=8) but significantly reduced in AAV9-GFP-treated age-matched control mice (n=8; *P<0.01 and #P<0.01).
Discussion
On the basis of the contradictory reports about the pathogenicity of specific sequence variants in the Sgcd gene,10, 11, 13 the existence of δ-sarcoglycan-associated autosomal dominant DCM remained questionable. The aim of our study was to investigate a possible relationship between the Sgcd gene missense mutation p.S151A and autosomal dominant DCM. In our heterozygous S151A knock-in mice, only mild symptoms of cardiomyopathy became obvious: minor sarcolemmal damage and a trend toward an increased heart to body weight suggested a rather low pathogenic potential of the p.S151A missense mutation to cause cardiomyopathy. Systemic transfer of the Sgcd-S151A cDNA using AAV9 vectors resulted in reconstitution of the sarcoglycan complex and preservation of cardiac function in cardiomyopathic Sgcd-deficient mice similar to treatment with wild-type Sgcd cDNA. However, although δ-sarcoglycan was effectively integrated into the sarcolemma, AAV9-mediated gene expression of the Sgcd S151A cDNA was not able to prevent histopathology in Sgcd-deficient mice completely suggesting a mild pathogenic potential of this variant as a cause of cardiomyopathy.
Transgenic mouse models have been used for almost two decades to elucidate the role of mutations associated with cardiomyopathies.21 Mice overexpressing the p.S151A missense mutation in the Sgcd gene have been shown to develop severe cardiomyopathy at young age with an enhanced lethality depending on the expression level of the mutant protein.12 This is in line with transgenic mice that develop cardiomyopathy associated with myopathy because of overexpression of wild-type Sgcg cDNA.22
Transgenic mouse lines are a common tool to investigate functional properties of mutant proteins. However, the expression level of the transgene is influenced by the proximity of the integration site and observed effects may depend on the abundance of expression. Targeted insertion of a mutation by homologous recombination, instead, may generate mouse models that reflect the clinical phenotypes more precisely.23
Thus, by homologous recombination, we generated a knock-in mouse model of the Sgcd gene p.S151A missense mutation to investigate whether this variant can cause cardiomyopathy. Heterozygous S151A knock-in mice developed a rather mild phenotype of cardiomyopathy. A slight but significant increase in heart to body weight suggested mild cardiac enlargement. It should be considered that heart to body weight calculations are prone to systematical errors and generally should be interpreted with caution. However, S151A knock-in mice show normal muscle mass and their absolute heart weights were increased compared with wild-type mice (data not shown). The underlying structural explanation could not be defined as there was no left ventricular dilatation and cross-sectional size of cardiomyocytes in hearts of S151A knock-in mice was not different from wild-type hearts (data not shown). Preserved myocardial contractility, the absence of histopathological alterations and normal life expectancy in these mice may explain why cardiac pathology might be missed by routine cardiac assessments in particular in young patients carrying the p.S151A missense mutation.13
Our TaqMan assay revealed that the mutant allele is expressed at approximately 4% of the wild-type allele. Such low levels raise the question whether haploinsufficiency contributes to the mild cardiac phenotype either by reduced expression of the mutant allele because of the design of the gene targeting plasmid (presence of the neoR cassette) or decreased stability of the mutant mRNA itself. As we had not sufficient amounts of cardiac biopsy material to address this question in patients, we chose to study the effect of haploinsufficiency in heterozygous δ-sarcoglycan-deficient mice. As heterozygous Sgcd+/− mice showed no signs of cardiomyopathy, haploinsufficiency per se does not completely explain the mild cardiac phenotype.
The absence of homozygous offspring carrying the p.S151A mutant δ-sarcoglycan on both alleles indicated early effects of the mutant protein leading to embryonic lethality before E10.5dpc. However, mice heterozygous for the mutant p.S151A allele most suitably reflect the heterozygous situation of our patients and may thus represent the human phenotype. A gain-of-function of the p.S151A allele – especially in early embryogenesis – could explain embryonic lethality in contrast to homozygous knock-out mice, which appear to be not affected in embryonic development. Further studies including quantitation of protein expression during embryonic development are necessary to finally answer this question.
δ-Sarcoglycan is essential for the stability of the sarcoglycan complex and its absence results in a loss of the other sarcoglycans from the plasma membrane. Dissociation of the DGC ultimately destabilizes sarcolemmal integrity leading to contraction-induced cell damage.24 Sgcd-deficient mice, in this regard, develop early cardiomyopathy and muscular dystrophy with signs of progressive necrosis, inflammation, fibrosis and regeneration and defective myocardial contractility.8, 9, 14 We previously reported that this process can be reduced by systemic transfer of the Sgcd gene using AAV-9 vectors transcriptionally targeted to the heart of Sgcd-deficient mice.14 AAV-9 vectors were used for cardiac gene transfer because they enable long-term and homogenous gene expression in the murine heart after intravenous vector injection.25, 26, 27, 28 Our previous study suggested that wild-type δ-sarcoglycan protein is produced sufficiently to restore the sarcoglycan complex in the plasma membrane and to maintain cardiac function in Sgcd-deficient mice. As expression of the mutant sarcoglycan in our knock-in model appeared to be low, we further compared AAV-mediated expression of the Sgcd-S151A mutant with the wild-type protein in Sgcd-deficient mice. AAV-mediated gene transfer of the Sgcd-S151A mutation resulted in a comparable α- and δ-sarcoglycan expression than the wild-type cDNA. Nevertheless, a mild cardiac histopathology occurred. Although the AAV gene transfer approach differs from the transgenic model, where the mutant sarcoglycan is severely underexpressed, development of fibrosis after gene transfer of the Sgcd-S151A mutation suggests a distinct pathological effect of the mutation.
On the molecular level, a transversion from thymidine 451 to guanine in exon 6 of the Sgcd gene replaces serine at codon 151 to alanine (p.S151A) changing a polar to a non-polar amino acid. Consistent with this a conformational change of δ-sarcoglycan might occur. The location of the S151A δ-sarcoglycan mutation at the extracellular domain may induce mild effects on sarcoglycan complex assembly10 and rather not affect sarcolemmal integrity. However, an additional contribution of the reduced expression of the δ-sarcoglycan mutation to the mild cardiac phenotype as observed in our transgenic model cannot be ruled out. In addition, substitution by increased expression of other sarcoglycans may further influence penetrance in our mouse model or in patients carrying the S151A mutation.29 Further studies are necessary to address the causes and role of decreased expression of mutant δ-sarcoglycan in detail.
In summary, our results suggest that the Sgcd gene p.S151A missense mutation can cause a mild, subclinical phenotype of cardiomyopathy unnoticed by routine cardiac examinations of patients carrying this mutation. In addition, this study underlines that an AAV-based cardiac gene transfer approach might be valuable to examine the disease-causing potential of distinct mutations, particularly when the resultant clinical phenotypes are inconclusive.
References
Heydemann A, McNally EM : Consequences of disrupting the dystrophin-sarcoglycan complex in cardiac and skeletal myopathy. Trends Cardiovasc Med 2007; 17: 55–59, Review.
Lapidos KA, Kakkar R, McNally EM : The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 2004; 94: 1023–1031.
Yoshida T, Pan Y, Hanada H, Iwata Y, Shigekawan M : Bidirectional signaling between sarcoglycans and the integrin adhesion system in cultured L6 myocytes. J Biol Chem 1998; 273: 1583–1590.
Yoshida M, Hama H, Ishikawa-Sakurai M et al: Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum Mol Genet 2000; 9: 1033–1040.
Zatz M, de Paula F, Starling A, Vainzof M : The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 2003; 13: 532–544.
Sandonà D, Betto R : Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects. Expert Rev Mol Med 2009; 11: e28.
Politano L, Nigro V, Passamano L et al: Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul Disord 2001; 11: 178–185.
Hack AA, Lam MY, Cordier L et al: Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci 2000; 113: 2535–2544.
Coral-Vazquez R, Cohn RD, Moore SA et al: Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999; 98: 465–474.
Tsubata S, Bowles KR, Vatta M et al: Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest 2000; 106: 655–662.
Kärkkäinen S, Miettinen R, Tuomainen P et al: A novel mutation, Arg71Thr, in the delta-sarcoglycan gene is associated with dilated cardiomyopathy. J Mol Med 2003; 81: 795–800.
Heydemann A, Demonbreun A, Hadhazy M, Earley JU, McNally EM : Nuclear sequestration of delta-sarcoglycan disrupts the nuclear localization of lamin A/C and emerin in cardiomyocytes. Hum Mol Genet 2007; 16: 355–363.
Bauer R, Hudson J, Müller HD et al: Does δ-sarcoglycan-associated autosomal dominant cardiomyopathy exist? Eur J Hum Genet 2009; 17: 1148–1153.
Goehringer C, Rutschow D, Bauer R et al: Prevention of cardiomyopathy in δ-sarcoglycan knock-out mice after systemic transfer of targeted adeno-associated viral vectors. Cardiovasc Res 2009; 82: 404–410.
Zang P, Li MZ, Elledge SJ : Towards genetic genomeprojects: genomic library screening and gene-targeting vector construction in a single step. Nat Genet 2002; 30: 31–39.
Liu P, Jenkins NA, Copeland NG : A highly efficient recombineering-based method for generating conditional knockout mutations. Genome 2003; 13: 476–484.
Geiger SK, Bär H, Ehlermann P et al: Incomplete nonsense-mediated decay of mutant lamin A/C mRNA provokes dilated cardiomyopathy and ventricular tachycardia. J Mol Med 2008; 86: 281–289.
Müller OJ, Schinkel S, Kleinschmidt JA, Katus HA, Bekeredjian R : Augmentation of AAV-mediated cardiac gene transfer after systemic administration in adult rats. Gene Ther 2008; 15: 1558–1565.
Hauswirth WW, Lewin AS, Zolotukhin S, Muzyczka N : Production and purification of recombinant adeno-associated virus. Methods Enzymol 2000; 316: 743–761.
Jung D, Duclos F, Apostol B : Characterization of delta-sarcoglycan, a novel component of the oligomeric sarcoglycan complex involved in limb-girdle muscular dystrophy. J Biol Chem 1996; 271: 32321–32329.
Morimoto S : Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res 2008; 77: 659–666.
Zhu X, Hadhazy M, Groh ME, Wheeler MT, Wollmann R, McNally EM : Overexpression of gamma-sarcoglycan induces severe muscular dystrophy. Implications for the regulation of Sarcoglycan assembly. J Biol Chem 2001; 276: 21785–21790.
Haruyama N, Cho A, Kulkarni AB : Overview: engineering transgenic constructs and mice. Curr Protoc Cell Biol 2009;, Unit 19.10, doi:10.1002/0471143030.cb1910s42.
Danialou G, Comtois AS, Dudley R et al: Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J 2001; 15: 1655–1657.
Müller OJ, Leuchs B, Pleger ST : Improved cardiac gene transfer by transcriptional and transductional targeting of adeno-associated viral vectors. Cardiovasc Res 2006; 70: 70–78.
Müller OJ, Katus HA, Bekeredjian R : Targeting the heart with gene therapy-optimized gene delivery methods. Cardiovasc Res 2007; 73: 453–462.
Inagaki K, Fuess S, Storm TA et al: Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 2006; 14: 45–53.
Pacak CA, Mah CS, Thattaliyath BD et al: Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res 2006; 99: e3–e9.
Gouveia TL, Kossugue PM, Paim JF et al: A new evidence for the maintenance of the sarcoglycan complex in muscle sarcolemma in spite of the primary absence of d-SG protein. J Mol Med 2007; 85: 415–420.
Acknowledgements
We thank Barbara Leuchs and the DKFZ vector core production unit for generating high titer AAV vector stocks. In addition, we thank Ulrike Gärtner (University of Heidelberg) for skillful intravenous injections of AAV vectors and Ursel Warncke (Max-Planck Institute for Medical Research, Heidelberg) for homologous recombination of ES cells. We further thank Stefan Selbert, PhD, (Polygene AG, Rümlang, Switzerland) for help in analyzing homologous recombination and the Nikon Center Heidelberg for support in microscopy. This work was supported by a grant of the Deutsche Forschungsgemeinschaft (MU 1654/3-2 to OJM); and the DZHK (German Centre for Cardiovascular Research) and the BMBF (German Ministry of Education and Research) to OJM and HAK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Rutschow, D., Bauer, R., Göhringer, C. et al. S151A δ-sarcoglycan mutation causes a mild phenotype of cardiomyopathy in mice. Eur J Hum Genet 22, 119–125 (2014). https://doi.org/10.1038/ejhg.2013.97
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.97
Keywords
This article is cited by
-
The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction
Communications Biology (2022)

{kind=link}
{kind=link}