
Abstract
Nance-Horan Syndrome (NHS) or X-linked cataract-dental syndrome is a disease of unknown gene action mechanism, characterized by congenital cataract, dental anomalies, dysmorphic features and, in some cases, mental retardation. We performed linkage analysis in a Tunisian family with NHS in which affected males and obligate carrier female share a common haplotype in the Xp22.32-p11.21 region that contains the NHS gene. Direct sequencing of NHS coding exons and flanking intronic sequences allowed us to identify the first missense mutation (P551S) and a reported SNP-polymorphism (L1319F) in exon 6, a reported UTR–SNP (c.7422 C>T) and a novel one (c.8239 T>A) in exon 8. Both variations P551S and c.8239 T>A segregate with NHS phenotype in this family. Although truncations, frame-shift and copy number variants have been reported in this gene, no missense mutations have been found to segregate previously. This is the first report of a missense NHS mutation causing NHS phenotype (including cardiac defects). We hypothesize also that the non-reported UTR–SNP of the exon 8 (3′-UTR) is specific to the Tunisian population.
Similar content being viewed by others


Introduction
Nance-Horan Syndrome (NHS; OMIM 302350) is an X-linked cataract-dental syndrome, characterized by bilateral congenital cataracts, dental anomalies, craniofacial abnormalities and, in some cases, mental retardation (MR).1, 2, 3 This syndrome is caused by mutations in the NHS gene (NHS) located on Xp22.13.4
Ophthalmological findings in affected males include bilateral severe congenital cataract involving the fetal nucleus and posterior Y suture, with variable zonular extensions into the posterior cortex.5, 6, 7, 8 Heterozygous females often manifest similar, but less severe features than affected males.5, 9 Approximately 30% of affected males display MR and behavioral disturbance.2, 5
NHS was mapped to Xp21.1-p22.37, 8 and, more recently, was refined to a critical region of 1.3 Mb between STR markers DXS1195 and DXS999.9 We have independently confirmed the genetic interval Xp22.32-p11.21 by detailed analysis of a Tunisian family with NHS.
The NHS gene is alternatively spliced and encodes two major isoforms, NHS-A and NHS-1A, implicated in the pathogenesis of NHS and containing a functional WAVE homology domain (WHD) in the N-terminus.10, 11 The NHS-A is able to associate with the cell membrane and involved in cell–cell contact.12 NHS protein is involved in the regulation of actin remodeling and cell morphology.11
Moreover, it is of importance in the limbic system, NHS protein which is of interest, given the range of neuropsychological abnormalities, including MR, autism, aggression, anxiety, stereotypical behavior and mood disturbance2, 5 reported in some males affected with NHS and present in our studied patients.
To date, 18 pathogenic mutations have been identified in 4 of the 10 coding exons of the NHS gene, all of which are predicted to result in a truncated NHS protein.4, 13, 14, 15, 16, 17, 18
Our present study revealed, for the first time, a missense mutation in NHS genes’ exon 6 identified in the three studied brothers and a reported SNP was revealed in the same exon. We identified also two other variations in exon 8: a novel UTR–SNP in 3′-UTR, which is predicted to be specific to the Tunisian population and a reported one.
Methods
Subjects and sample collection
Six members of an NHS family have been enrolled in a genetic research program through the laboratory of Human Genetics (Faculté de Médecine de Tunis, Tunisia). The family was referred to the Department of Congenital and Hereditary Disorders at the Charles Nicolle Hospital (Tunis, Tunisia), because of the three affected brothers with signs of NHS, including congenital cataract, MR, dysmorphic features, dental anomalies and cardiac defect in one patient. The three patients were followed by ophthalmologists, because of severe bilateral cataract clinically suspected since birth. They had undergone cataract extraction early in life, before the age of 12 years.
Peripheral blood samples were taken from the three affected patients, their two healthy siblings (brother and sister) and their mother. Informed consent was obtained from affected and healthy family members.
A total of 50 control individuals, age from 20 to 50 years, were selected; they include 22 males and 28 females with no sign of MR, neither cataract or cardiac defect, nor dental anomalies.
Genomic DNA was extracted from peripheral blood leukocytes by the standard proteinase-K extraction consisting of: lysis of red blood cells by RBC Lysis Buffer (155 mM NH4Cl, 10 mM KHCO3, 0.5 EDTA, pH 7.5) and white blood cells by a WBC Lysis Buffer (1 mM Na-EDTA, 5 mM Tris HCl pH 7.5), treatment of the lysate with a mixture of detergent composed of SDS (sodium dodecyl sulfate or sacrosyl and proteinase K) in order to liberate the DNA and digest the associated proteins, precipitation of the DNA in the form of filaments by absolute ethanol and finally dilution of the DNA in T10E1 buffer (Tris 10 mM, EDTA 0.1–1 mM) and stored in 10 ml vacuum tube sterile containing 100 μl of 0.1 M EDTA.K3.
Genotyping
Polymerase chain reaction (PCR)-based microsatellite markers genotyping were performed by using 18 dinucleotide fluorescent microsatellites on the X-chromosome at 10-cM intervals (panel 28 of the Prism Linkage Mapping Set, ver.2.5; Applied Biosystems (ABI), Foster City, CA, USA). Genotyping was performed on a genetic analyser (PRISM 3130; ABI) with accompanying software (GeneScan; ABI).
Linkage analysis
Two-point LOD scores were calculated using the MLINK program of the LINKAGE package (ver. 4.1P; http://www.hgmp.mrc.ac.uk; provided in the public domain by the Human Genome Mapping Project Resources Centre, Cambridge, UK).19, 20 Analysis assumed an X-linked recessive transmission with complete penetrance.
Molecular analysis
Candidate gene NHS (isoform A) was screened for mutations and variations. The gene sequence was retrieved from the UCSC Human Genome Browser (http://genome.ucsc.edu/; NHS: NM_198270). Suitable primers for PCR amplification of exons and flanking regions sequences of NHS were designed using Primer 3 (Primer 3-http://frodo.wi.mit.edu/primer3/).
PCR was performed by using 400 ng of DNA template, 20 pmol each of forward and reverse primers (Biomatik, Cambridge, Canada), 1.5 units of Taq DNA polymerase recombinant (Invitrogen, Carlsbad, CA, USA) and 1.25 mM dNTPs (Promega, Madison, WI, USA) in a total volume of 50 μl. PCR consisted on 35 cycles and was carried out in an automated thermal cycle GeneAmp PCR System 9700 (ABI) under the following conditions: initial denaturation at 95 °C for 5 min and denaturation at 95 °C for 30 s, annealing at 52–60 °C for 30 s and elongation at 72 °C for 30 s, followed by one cycle of final extension at 72 °C for 7 min.
The amplified products were purified (Wizard SV Gel and PCR Clean-Up System Kit; Promega) and sequenced (Big Dye Terminator Cycle Sequencing Ready Reaction; DNA Sequencing Kit; ABI PRISM 3130; ABI) in the forward and reverse directions. Sequencing results were visualized and data were computer analyzed using Sequencing Analysis 5.2 and SeqScape softwares (ABI).
Comparing between normal protein and a mutated one
In order to compare the sequence of a normal protein with a mutated one we used the SMART site (Simple Modular Architecture Research Tool: http://smart.embl-heidelberg.de/) that allowed us to identify the differences between proteins’ domains.
Results
Clinical and biological investigations
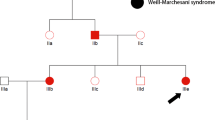
The three affected patients showed clinical presentations consistent with typical features of NHS shown in Table 1, but characteristic screwdriver blade-shaped incisors were not present. They had bilateral cataract involving the nucleus, total nuclear type with microphthalmia. None of them presented microcornea, nystagmus or strabismus. They showed moderate to severe MR; patients III14 and III15 (Figure 1a) had walked and spoken at 3 years. However, their affected brother III16 is intellectually better; he had walked and spoken at 2 years. Their neurological examination showed ligamentous laxity of the fingers, stereotyped hands and asymmetric bodies. Moroever, we sign up neonatal hypotonia and cardiac anomaly for patient III15.

(a) Pedigree of the family. Filled squares denote affected males showing features of Nance-Horan syndrome. (b) Haplotypes of the Xp22.32-Xp11.21 region. The shared haplotype between the three patients. ?, undetermined allele.
Karyotyping with R-banding revealed normal karyotypes: 46,XY (600 bands resolution) and normal metabolic screening, including normal Fehling reactions and normal thin layer chromatography of reducing sugars, normal plasmatic amino acid and urine organic acid chromatography. Magnetic resonance imaging of the brain were normal for the three screened brothers. Patients were born from healthy, consanguineous parents, the father II8 was dead after a cancer disease. On the basis of the pedigree of this family (Figure 1a), we suspected an X-linked transmission of congenital cataract with recessive inheritance.
Linkage analysis
Segregation analysis using the 18 polymorphic markers on the X-chromosome within 10-cM region allowed us to exclude implication in the NHS phenotype of most regions of the X-chromosome, except the region Xp22.32-p11.21, between the markers DXS1060 and DXS993 (Figure 1b). At this region, affected males were found to share a haplotype spanning about 50-Mb and the highest two-point LOD score within the interval was 0.90 (Table 2), assuming recessive inheritance. This region contains more than 10 genes implicated in non-syndromic forms of X-linked MR and a unique gene involved in X-linked congenital cataract, the NHS gene. For this reason, we considered the NHS gene as candidate for the studied family.
Mutational screening
All eight exons and the flanking regions of the NHS gene were screened for mutations. Four variations were detected in all three affected brothers, in a hemizygous trait, including a non reported missense mutation, c. 1989C>T (C reference allele), resulting in conversion of proline to serine: p. P551S (Figure 2a), and a reported polymorphism, c. 4293C>T (C reference allele), resulting in conversion of leucine to phenylalanine: p.L1319F; GenBank accession number dbSNP3747295 (http://www.ncbi.nlm.nih.gov), in exon 6. The two other variations are located in 3′-UTR exon8: a reported UTR–SNP, c. 7422C>T (C reference allele); GenBank accession number dbSNP60285340 and a novel one, c. 8239T>A (T reference allele) (Figure 2a).

Sequence chromatogram showing: (a) missense mutation in exon 6, c.1989C>T resulting in p.P551S in the three affected males (ii), their carrier mother (i) and normal sequence (iii) in two non-affected patients and 50 control individuals. (b) Non-reported UTR–SNP in exon 8, c. 8239 T>A in the three affected brothers (ii), their carrier mother (i) and normal sequence in two normal patients and 50 control individuals (iii).
The two novel variations in exon 6 (c. 1989C>T) and 3′-UTR exon 8 (c. 8239T>A) were absent in the 78 control chromosomes, suggesting the sequences changes in the two exons as a disease-causing, co-segregating mutation, which highlighted the correlation genotype–phenotype.
Moreover, the alignment of the sequence comprising the exon 6′variation, c. 1989C>T, with eight tested species proved that this substitution is conserved in all of them (Figure 3).

NHS protein sequences in nine vertebrates showing conservation of the amino-acid proline in position 551 in human NHS protein. (http://genome.ucsc.edu/).
This missense mutation, p. P551S, contributes to the appearance of a novel low complexity region. In fact, the normal NHS protein counts 12 low complexity region serine rich, whereas the mutated protein engendered by the missense mutation counts 13 (Figure 4); the novel complexity region found in the mutated protein is a serine-rich domain with tandem-repeat insertion (Figure 5).

Comparison in NHS protein structure between normal and mutated protein showing the additional LcR domain (grey-colored) in mutated protein. (http://smart.embl-heidelberg.de/) The position of the novel LcR is counted from the amino acid 1.

Mutated NHS protein sequence showing LcR domains (in rectangles) due to missense mutation p.P551S (the novel LcR is indicated in bold). Open rectangle indicate that the LcR is continued in the second line. The sequences of the LcRs are determined in the SMART site (http://smart.embl-heidelberg.de/).
Discussion
There are many inherited disease syndromes associated with congenital cataracts;21 among it is the NHS, which is an X-linked disorder associating mainly X-linked cataract to MR, dysmorphic features and dental anomalies.8
By linkage analysis, we localized the disease region near the NHS locus with two-point LOD score of 0.90, spanning the region between DXS1060 and DXS991 in which a novel gene, called NHS, was identified by positional cloning approach.4
NHS is a large gene that encodes a 1630-amino-acid major isoform A protein, 1335-amino-acid minor isoform B protein, which begins translation in exon 4,4 and 1651-amino-acid isoform 1A that is transcribed from exon 1 like the NHS-A. These two isoforms (A and 1A) differ in the presence or absence of exon 3a.11 NHS has an important regulatory role in the development of ocular, craniofacial and neural tissues. It has been demonstrated that NHS protein is expressed in midbrain, lens, tooth and retina.4 The fact that the NHS gene is highly conserved across vertebrate species supports the vital role of the gene in development.4
This is the first report of a missense mutation (p.P551S) identified in protein NHS. It was detected in the three affected males, their carrier mother (heterozygous) and was not detected in healthy brother and sister and in 50 control individuals; in addition, this substitution is conserved in eight tested species (Figure 3), confirming the sequence change as a disease-causing, cosegregating mutation. This mutation is the first missense mutation involving the NHS gene. All the other 15 reported mutations are nonsense mutations or frame-shift mutations leading to protein truncation or the complete absence of the protein, available in Human Gene Mutation database HGMD (http://www.hgmd.cf.ac.uk/) and in Mental Retardation database LOVD (http://grenada.lumc.nl/LOVD2/MR/variants). The family reported here, was addressed first for intellectual disability, we re-examined the family and noted that the clinical severity of other features of NHS was either mild or varied highly in some of the members; the phenotype in this family is less severe than those reported in the families, where truncating mutations were identified. This may be due to the fact that this missense mutation engenders a simple conversion of a proline to serine; the proline is a hydrophobic amino acid, whereas the serine is a hydrophilic one.
NHS protein may function as a key scaffold protein which controls the balance between cell adhesion and motility by coordinating the function of multiple regulatory pathways important for actin stability and plasticity.11 All mutations causing an NHS phenotype are null mutations predicted to produce no functional protein; most of them affected the WHD domain, localized on the N-terminal of NHS protein.11 In fact, the loss of NHS isoforms (NHS-A and NHS-1A) containing a functional WHD causes NHS.4, 13 But the p.P551S mutation identified in the studied family did not affect the WHD domain, but the serine-rich domain. It contributes to the appearance of a novel low complexity region (Figure 4); low-complexity regions are defined as fragments of sequences with non-random distribution of restricted number of residues. These low-complexity sequences occur often and in many (unrelated) types proteins and consist of repetitive short fragments (for example, leucine-rich domains or poly-alanine tracts).22, 23 In addition, serine is a hydrophilic amino acid; hydrophilic amino acids are typically found on protein surfaces exposed to an aqueous environment. The fact that the new low complexity serine-rich region is found in the middle of the protein may change the biochemical and structural properties and lead to functional impairment of the NHS protein and engender the NHS in the family reported here. In fact, in our NHS patients, developmental delay was observed in all studied three siblings, which we propose due to modifier effects affecting the serine-rich domain and influencing phenotype. Interestingly, we uggest that this missense mutation, causing disease, leads to moderate NHS phenotype.
We detected also a novel UTR–SNP in 3′-UTR exon8 (c.8239 T>A) in the three affected males (hemizygous) and their carrier mother (heterozygous). This variation was absent in their healthy brother and sister and 50 control individuals (22 males, 28 females); otherwise, this UTR–SNP was absent in 78 chromosomes confirming the association between genotypes and the studied phenotype (NHS ) for this Tunisian family. The novel UTR–SNP was not reported neither in NCBI reference assembly (http://www.ncbi.nlm.nih.gov/), nor in Ensembl Human Gene (http://www.ensembl.org/Homo-Sapiens), so we suggest that it could be specific to the Tunisian population.
Moreover, the linked region Xp22.32-p11.21, identified by genotyping, contains 10 other genes implicated in non-syndromic forms of X-linked MR: AP1S2, ARX, ZNF674, ZNF41, ZNF81, FTSJ1, PQBP1, SYP, SHROOM4 (or KIAA1202) and JARID1C. No one of these has been reported responsible for congenital cataract neither contributing to the development of the eye. As a result, none of these 10 genes could be responsible for the phenotype (X-linked congenital cataract associated to MR) in the studied family. In this region, only the NHS gene is expressed in the eye (lens and retina),4 and its’ mutation was described leading to X-linked congenital cataract.5 These findings confirm that the novel missense mutation P551S, identified for the first time, contributed alone or in conjunction with the novel 3′UTR–SNP c. 8239T>A to the mild NHS phenotype in our patients.
In our studied family, one of the two affected patients (III15) presented X-linked congenital cataract and moderate features of NHS (MR, oligodentia, facial dysmorphism), with a cardiac anomaly (cardiac surgery, disposed a valve). In fact, in 2002, a family was reported by Francis et al.24 as having X-linked congenital cataract associated with cardiac anomalies, mapping to the region Xp22.13. In 2003, Burdon et al.4 confirmed the same localization (Xp22.13) for the NHS gene that was found to be expressed in the heart. Recently, Coccia and co-workers25 reported four of six affected males, belonging to a same family, with congenital heart defects with no typical features of NHS locus. By sequence analysis of NHS gene, they did not identify any mutation, but with using X-chromosome array they revealed copy number variation (segmental duplication–triplication) leading to altered transcriptional regulation of the protein NHS, which supports the hypothesis that aberrant transcription of the NHS gene is responsible for congenital heart defects in X-linked congenital cataract family. Taking these results further, we hypothesize that NHS could be responsible for the heart defect also in our patient.
Our report is the first report of a missense mutation within NHS gene in a Tunisian family with X-linked congenital cataract and moderate features of NHS syndrome. We had also identified a novel 3′ UTR–SNP that could be specific to the Tunisian population.
As perspective, we aimed to study the effect of such identified missense mutation and the 3′UTR–SNP in NHS protein structure by mouse mutagenesis.
Accession codes
References
Hibbert S : A previously unreported association between Nance-Horan syndrome and spontaneous dental abscesses. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2005; 99: 207–211.
Toutain A, Ayrault AD, Moraine C : Mental retardation in Nance-Horan syndrome: clinical and neuropsychological assessment in four families. Am J Med Genet 1997; 71: 305–314.
Ding X, Patel M, Herzlich AA, Sieving PC, Chan CC : Ophthalmic pathology of Nance-Horan syndrome: case report and review of the literature. Ophthalmic Genet 2009; 30: 127–135.
Burdon KP, McKay JD, Sale MM et al: Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am J Hum Genet 2003; 73: 1120–1130.
Walpole IR, Hockey A, Nicoll A : The Nance-Horan syndrome. J Med Genet 1990; 27: 632–634.
Lewis RA, Nussbaum RL, Stambolian D : Mapping X-linked ophthalmic diseases. IV. Provisional assignment of the locus for X-linked congenital cataracts and microcornea (the Nance-Horan syndrome) to Xp22.2-p22.3. Ophthalmology 1990; 97: 110–120.
Stambolian D, Lewis RA, Buetow K, Bond A, Nussbaum R : Nance-Horan syndrome: localization within the region Xp21.1-Xp22.3 by linkage analysis. Am J Hum Genet 1990; 47: 13–19.
Bixler D, Higgins M, Hartsfield J : The Nance-Horan syndrome: a rare X-linked ocular-dental trait with expression in heterozygous females. Clin Genet 1984; 26: 30–35.
Zhu D, Alcorn DM, Antonarakis SE et al: Assignment of the Nance-Horan syndrome to the distal short arm of the X chromosome. Hum Genet 1990; 86: 54–58.
Toutain A, Dessay B, Ronce N et al: Refinement of the NHS locus on chromosome Xp22.13 and analysis of five candidate genes. Eur J Hum Genet 2002; 10: 516–520.
Brooks SP, Coccia M, Tang HR et al: The Nance-Horan syndrome protein encodes a functional WAVE homology domain (WHD) and is important for co-ordinating actin remodelling and maintaining cell morphology. Hum Mol Genet 2010; 19: 2421–2432.
Sharma S, Koh KS, Collin C et al: NHS-A isoform of the NHS gene is a novel interactor of ZO-1. Exp Cell Res 2009; 14: 2358–2372.
Brooks SP, Ebenezer ND, Poopalasundaram S, Lehmann OJ, Moore AT, Hardcastle AJ : Identification of the gene for Nance-Horan syndrome (NHS). J Med Genet 2002; 41: 768–771.
Ramprasad VL, Thool A, Murugan S et al: Truncating mutation in the NHS gene: phenotypic heterogeneity of Nance-Horan syndrome in an Asian Indian family Invest. Ophthalmol Vis Sci 2005; 46: 17–23.
Florijn RJ, Loves W, Maillette de Buy Wenniger-Prick LJ, Mannens MM et al: New mutations in the NHS gene in Nance-Horan Syndrome families from the Netherlands. Eur J Hum Genet 2006; 14: 986–990.
Huang KM, Wu J, Brooks SP, Hardcastle AJ, Lewis RA, Stambolian D : Identification of three novel NHS mutations in families with Nance-Horan syndrome. Mol Vis 2007; 13: 470–474.
Reches A, Yaron Y, Burdon K et al: Prenatal detection of congenital bilateral cataract leading to the diagnosis of Nance-Horan syndrome in the extended family. Prenat Diagn 2007; 27: 662–664.
Sharma S, Burdon KP, Dave A et al: Novel causative mutations in patients with Nance-Horan syndrome and altered localisation of the mutant NHS-A protein isoform. Mol Vis 2008; 14: 1856–1864.
Cottingham RW, Idury RM, Schaffer AA : Faster sequential genetic linkage computations. Am J Hum Genet 1993; 53: 252–263.
Schaffer AA, Gupta SK, Shriram K, Cottingham RW : Avoiding recomputation in linkage analysis. Hum Hered 1994; 44: 225–237.
He W, Li S : Congenital cataract: gene mapping. Hum Genet 2000; 106: 1–13.
Bannen RM, Bingman CA, Philips GN : Effect of low-complexity regions on protein structure determination. J Struct Funct Genomics 2007; 8: 217–226.
Brocchieri L : Low-complexity regions in Plasmodium proteins: in a search of a function. Genome Res 2009; 11: 195–197.
Francis PJ, Berry V, Hardcastle AJ, Maher ER, Moore AT, Bhattacharya SS : A locus for isolated cataract on human Xp. J Med Genet 2002; 39: 105–109.
Coccia M, Brooks SP, Webb TR et al: X-linked cataract and Nance-Horan syndrome are allelic disorders. Hum Mol Genet 2009; 18: 2643–2655.
Acknowledgements
We thank all the patients and their family members for participating in the project. This study was supported by the Tunisian ministry of higher education and scientific research (Laboratory of Human Genetics, Faculté de Médecine de Tunis and Congenital and Hereditary service of Charles Nicolle's Hospital).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Chograni, M., Rejeb, I., Jemaa, L. et al. The first missense mutation of NHS gene in a Tunisian family with clinical features of NHS syndrome including cardiac anomaly. Eur J Hum Genet 19, 851–856 (2011). https://doi.org/10.1038/ejhg.2011.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.52
Keywords
This article is cited by
-
Clinical and genetic findings in patients with congenital cataract and heart diseases
Orphanet Journal of Rare Diseases (2021)
-
Allelic and dosage effects of NHS in X-linked cataract and Nance–Horan syndrome: a family study and literature review
Molecular Cytogenetics (2021)
-
A novel small deletion in the NHS gene associated with Nance-Horan syndrome
Scientific Reports (2018)
-
Congenital diaphragmatic hernia as a part of Nance–Horan syndrome?
European Journal of Human Genetics (2018)
-
A non-syndromic intellectual disability associated with a de novo microdeletion at 7q and 18p, microduplication at Xp, and 18q partial trisomy detected using chromosomal microarray analysis approach
Molecular Cytogenetics (2014)
