
Key Points
-
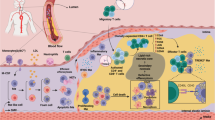
Atherothrombotic vascular disease (ATVD) is the leading cause of death in the industrialized world, and this problem is growing owing to the increase in obesity and insulin resistance worldwide. ATVD develops as a macrophage-dominant maladaptive inflammatory response to subendothelial lipoproteins.
-
A fundamental aspect of this maladaptive inflammatory response is a failure to resolve inflammation, which normally involves the suppression of inflammatory cell influx, effective phagocytic clearance of apoptotic cells and promotion of inflammatory cell egress. The mechanism of failed inflammation resolution in atherosclerosis is not known, but it is likely to involve defective generation or action of anti-inflammatory cytokines (for example, interleukin-10), pro-resolution lipid mediators (for example, lipoxins) and transcription factors (for example, the liver X receptor family) that normally carry out this process.
-
In advanced atherosclerosis, there is continual recruitment of inflammatory monocytes, and the macrophages that differentiate from these monocytes in lesions may favour the classically activated (M1) subtype, which promote inflammation, over the alternatively activated (M2) subtype, which participate in inflammation resolution. Egress of inflammatory macrophages in advanced atherosclerotic lesions is also defective.
-
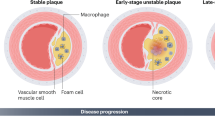
Macrophage apoptosis coupled with defective clearance of these apoptotic cells (efferocytosis) in advanced atherosclerotic lesions is a particularly important process because it leads to the generation of plaque necrosis, which is a key feature of the types of atherosclerotic lesions that cause ATVD. One key mechanism of macrophage apoptosis in this setting is a pathway in which the endoplasmic reticulum stress pathway known as the unfolded protein response, perhaps in combination with pattern recognition receptor activation, triggers Ca2+-dependent apoptosis.
-
The mechanisms of defective efferocytosis in advanced atheroma are not known, but may involve deficiency, dysfunction and/or competitive inhibition of receptors, ligands and other factors involved in apoptotic cell recognition and engulfment.
-
The ability to translate the complex process of plaque progression into an integrated molecular and cellular concept of defective inflammation resolution provides a useful way to understand how atherosclerosis leads to clinical disease and how plaque progression may be prevented by new therapeutic approaches.
Abstract
A key event in atherosclerosis is a maladaptive inflammatory response to subendothelial lipoproteins. A crucial aspect of this response is a failure to resolve inflammation, which normally involves the suppression of inflammatory cell influx, effective clearance of apoptotic cells and promotion of inflammatory cell egress. Defects in these processes promote the progression of atherosclerotic lesions into dangerous plaques, which can trigger atherothrombotic vascular disease, the leading cause of death in industrialized societies. In this Review I provide an overview of these concepts, with a focus on macrophage death and defective apoptotic cell clearance, and discuss new therapeutic strategies designed to boost inflammation resolution in atherosclerosis.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
References
Serhan, C. N. et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332 (2007). This review presents an outstanding summary of the principles of inflammation resolution, many of which apply to the concept of resolution failure in atherosclerosis.
Lawrence, T. & Gilroy, D. W. Chronic inflammation: a failure of resolution? Int. J. Exp. Pathol. 88, 85–94 (2007).
Martinez, F. O., Helming, L. & Gordon, S. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483 (2009).
[No authors listed]. 2007 NHLBI Morbidity and Mortality Chart Book. NHLBI Morbidity and Mortality Chartbook [online], (2007).
Grundy, S. M. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 89, 2595–2600 (2004).
Tabas, I., Williams, K. J. & Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116, 1832–1844 (2007).
Lusis, A. J. Atherosclerosis. Nature 407, 233–241 (2000).
Glass, C. K. & Witztum, J. L. Atherosclerosis: the road ahead. Cell 104, 503–516 (2001).
Tabas, I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol. 25, 2255–2264 (2005).
Schrijvers, D. M., De Meyer, G. R., Herman, A. G. & Martinet, W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc. Res. 73, 470–480 (2007). References 9 and 10 present the concept that efferocytosis, a key process in inflammation resolution, is defective in advanced atherosclerotic lesions and is an important factor in plaque necrosis.
Merched, A. J., Ko, K., Gotlinger, K. H., Serhan, C. N. & Chan, L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 22, 3595–3606 (2008). This paper provides evidence that lipid mediators of inflammation resolution derived from 12,15-lipoxygenase, including lipoxin A4, resolvin D1 and protectin D1, suppress atherosclerotic plaque progression in the Apoe−/− mouse model of atherosclerosis.
Randolph, G. J. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr. Opin. Lipidol. 19, 462–468 (2008).
Virmani, R., Burke, A. P., Kolodgie, F. D. & Farb, A. Vulnerable plaque: the pathology of unstable coronary lesions. J. Interv. Cardiol. 15, 439–446 (2002).
Swirski, F. K. et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc. Natl Acad. Sci. USA 103, 10340–10345 (2006).
Swirski, F. K. et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205 (2007). References 14 and 15 show that inflammatory monocytes continually enter atherosclerotic lesions even after inflammation has been long-standing. This finding supports the concept that a key process in inflammation resolution (eventual suppression of inflammatory cell influx) is defective in atherosclerosis.
Chawla, A. et al. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nature Med. 7, 48–52 (2001).
Odegaard, J. I. et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 (2007). Although not dealing directly with atherogenesis, this paper provides a link between the PPAR family of transcriptional activators and a key process in inflammation resolution, namely the alternative activation of macrophages.
Nofer, J. R. et al. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 115, 501–508 (2007).
Laurat, E. et al. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 104, 197–202 (2001).
Li, A. C. et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. J. Clin. Invest. 114, 1564–1576 (2004). This study showed that PPAR γ , a mediator of inflammation resolution and cholesterol efflux in atherosclerotic macrophages, has a beneficial effect on lesion progression.
Angeli, V. et al. Dyslipidemia associated with atherosclerotic disease systemically alters dendritic cell mobilization. Immunity 21, 561–574 (2004).
Llodra, J. et al. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl Acad. Sci. USA 101, 11779–11784 (2004). This paper provides in vivo evidence that macrophage egress, a key process in inflammation resolution, is defective in mouse atherosclerotic lesions but that it can occur in a setting in which regression of atherosclerosis is promoted.
Quah, B. J. & O'Neill, H. C. Maturation of function in dendritic cells for tolerance and immunity. J. Cell. Mol. Med. 9, 643–654 (2005).
Trogan, E. et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl Acad. Sci. USA 103, 3781–3786 (2006).
Mosser, D. M. & Zhang, X. Interleukin-10: new perspectives on an old cytokine. Immunol. Rev. 226, 205–218 (2008).
Serhan, C. N., Chiang, N. & Van Dyke, T. E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature Rev. Immunol. 8, 349–361 (2008).
Willoughby, D. A., Moore, A. R., Colville-Nash, P. R. & Gilroy, D. Resolution of inflammation. Int. J. Immunopharmacol. 22, 1131–1135 (2000).
Serhan, C. N. et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 206, 15–23 (2009).
Joseph, S. B., Castrillo, A., Laffitte, B. A., Mangelsdorf, D. J. & Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nature Med. 9, 213–219 (2003).
Huang, J. T. et al. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382 (1999).
Li, Y. et al. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signalling mechanism. J. Biol. Chem. 283, 34833–34843 (2008).
Lingnau, M., Hoflich, C., Volk, H. D., Sabat, R. & Docke, W. D. Interleukin-10 enhances the CD14-dependent phagocytosis of bacteria and apoptotic cells by human monocytes. Hum. Immunol. 68, 730–738 (2007).
Pinderski, L. J. et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient mice by altering lymphocyte and macrophage phenotypes. Circ. Res. 90, 1064–1071 (2002). This paper directly links the inflammation-resolving cytokine IL-10 with the key end point of plaque necrosis in mouse atherosclerosis.
Seljeflot, I., Hurlen, M., Solheim, S. & Arnesen, H. Serum levels of interleukin-10 are inversely related to future events in patients with acute myocardial infarction. J. Thromb. Haemost. 2, 350–352 (2004).
Wahl, S. M., Swisher, J., Cartney-Francis, N. & Chen, W. TGF-β: the perpetrator of immune suppression by regulatory T cells and suicidal T cells. J. Leukoc. Biol. 76, 15–24 (2004).
Huynh, M. L., Fadok, V. A. & Henson, P. M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41–50 (2002).
Frutkin, A. D. et al. TGF-β1 limits plaque growth, stabilizes plaque structure, and prevents aortic dilation in apolipoprotein e-null mice. Arterioscler. Thromb. Vasc. Biol. 29, 1251–1257 (2009).
Mallat, Z. et al. Inhibition of transforming growth factor-β signalling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 89, 930–934 (2001).
Lutgens, E. et al. Transforming growth factor-β mediates balance between inflammation and fibrosis during plaque progression. Arterioscler. Thromb. Vasc. Biol. 22, 975–982 (2002).
Bahekar, A. A., Singh, S., Saha, S., Molnar, J. & Arora, R. The prevalence and incidence of coronary heart disease is significantly increased in periodontitis: a meta-analysis. Am. Heart J. 154, 830–837 (2007).
Van Dyke, T. E. Resolution of inflammation-unraveling mechanistic links between periodontitis and cardiovascular disease. J. Dent. 37, S582–S583 (2009).
Peters-Golden, M. Putting on the brakes: cyclic AMP as a multipronged controller of macrophage function. Sci. Signal. 2, pe37 (2009).
Takayama, K. et al. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J. Biol. Chem. 277, 44147–44154 (2002).
Wall, E. A. et al. Suppression of LPS-induced TNF-α production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci. Signal. 2, ra28 (2009).
Babaev, V. R. et al. Macrophage EP4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metab. 8, 492–501 (2008).
Marathe, C. et al. The arginase II gene is an anti-inflammatory target of liver X receptor in macrophages. J. Biol. Chem. 281, 32197–32206 (2006).
Hong, C. & Tontonoz, P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr. Opin. Genet. Dev. 18, 461–467 (2008).
Castrillo, A., Joseph, S. B., Marathe, C., Mangelsdorf, D. J. & Tontonoz, P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J. Biol. Chem. 278, 10443–10449 (2003).
Venkateswaran, A. et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXRα. Proc. Natl Acad. Sci. USA 97, 12097–12102 (2000).
Tall, A. R. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J. Intern. Med. 263, 256–273 (2008).
Joseph, S. B. et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl Acad. Sci. USA 99, 7604–7609 (2002). This paper provides in vivo evidence linking LXR family members, mediators of inflammation resolution and cholesterol efflux in atherosclerotic macrophages, with a beneficial effect on lesion progression.
Levin, N. et al. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler. Thromb. Vasc. Biol. 25, 135–142 (2005).
Bradley, M. N. et al. Ligand activation of LXRβ reverses atherosclerosis and cellular cholesterol overload in mice lacking LXRα and apoE. J. Clin. Invest. 117, 2337–2346 (2007).
Rebe, C. et al. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor-α pathway increases the clearance of apoptotic cells by human macrophages. Circ. Res. 105, 393–401 (2009).
Gonzalez, N. et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 (2009).
Kockx, M. M. et al. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation 97, 2307–2315 (1998).
Liu, J. et al. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 25, 174–179 (2005).
Boesten, L. S. et al. Macrophage p53 controls macrophage death in atherosclerotic lesions of apolipoprotein E deficient mice. Atherosclerosis 88, 780–786 (2009).
Arai, S. et al. A role for the apoptosis inhibitory factor AIM/Spα/Api6 in atherosclerosis development. Cell Metab. 1, 201–213 (2005).
Wang, B. Y. et al. Regression of atherosclerosis: role of nitric oxide and apoptosis. Circulation 99, 1236–1241 (1999).
Bhatia, V. K. et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am. J. Pathol. 170, 416–426 (2007).
Tabas, I. Apoptosis and plaque destabilization: the role of macrophage apoptosis induced by cholesterol. Cell Death. Differ. 11, S12–S16 (2004).
Zinszner, H. et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 (1998).
Li, G. et al. Role of ERO1α-mediated stimulation of inositol 1, 4, 5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186, 783–792 (2009).
Timmins, J. M. et al. Calcium/calmodulin-dependent protein kinase II links endoplasmic reticulum stress with Fas and mitochondrial apoptosis pathways. J. Clin. Invest. 119, 2925–2941 (2009).
Seimon, T. & Tabas, I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J. Lipid Res. 50, S382–S387 (2009).
DeVries-Seimon, T. et al. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 171, 61–73 (2005).
Seimon, T. A., Obstfeld, A., Moore, K. J., Golenbock, D. T. & Tabas, I. Combinatorial pattern recognition receptor signalling alters the balance of life and death in macrophages. Proc. Natl Acad. Sci. USA 103, 19794–19799 (2006).
Rosenfeld, M. E. et al. Animal models of spontaneous plaque rupture: the holy grail of experimental atherosclerosis research. Curr. Atheroscler. Rep. 4, 238–242 (2002).
Feng, B. et al. Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc. Natl Acad. Sci. USA 100, 10423–10428 (2003).
Lim, W. S. et al. STAT1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation 117, 940–951 (2008).
Thorp, E. et al. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 9, 474–481 (2009). This paper provides in vivo evidence that the UPR effector CHOP has an important role in macrophage apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice.
Manning-Tobin, J. J. et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 29, 19–26 (2009).
Liang, C. P. et al. Increased CD36 protein as a response to defective insulin signalling in macrophages. J. Clin. Invest. 113, 764–773 (2004).
Han, S. et al. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 3, 257–266 (2006).
Seimon, T. A. et al. Macrophage deficiency of p38α MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J. Clin. Invest. 119, 886–898 (2009).
Gargalovic, P. S. et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26, 2490–2496 (2006).
Myoishi, M. et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 116, 1226–1233 (2007).
Sanson, M. et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ. Res. 104, 328–336 (2009).
Henson, P. M., Bratton, D. L. & Fadok, V. A. Apoptotic cell removal. Curr. Biol. 11, R795–R805 (2001).
De Lorenzo, B. H. et al. Macrophage suppression following phagocytosis of apoptotic neutrophils is mediated by the S100A9 calcium-binding protein. Immunobiology 3 Aug 2009 (doi:10.1016/j.imbio.2009.05.013).
Mallat, Z. et al. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 99, 348–353 (1999).
Schrijvers, D. M., De Meyer, G. R., Kockx, M. M., Herman, A. G. & Martinet, W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 25, 1256–1261 (2005). This study used an in situ assay to provide evidence that efferocytosis is defective in advanced human atherosclerotic lesions.
Ravichandran, K. S. & Lorenz, U. Engulfment of apoptotic cells: signals for a good meal. Nature Rev. Immunol. 7, 964–974 (2007).
Peter, C. et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J. Biol. Chem. 283, 5296–5305 (2008).
Gardai, S. J. et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through transactivation of LRP on the phagocyte. Cell 123, 321–334 (2005).
Zhou, Z. New phosphatidylserine receptors: clearance of apoptotic cells and more. Dev. Cell 13, 759–760 (2007).
Boisvert, W. A. et al. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler. Thromb. Vasc. Biol. 26, 563–569 (2006).
Ait-Oufella, H. et al. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation 115, 2168–2177 (2007).
Thorp, E., Cui, D., Schrijvers, D. M., Kuriakose, G. & Tabas, I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of Apoe−/− mice. Arterioscler. Thromb. Vasc. Biol. 28, 1421–1428 (2008).
Ait-Oufella, H. et al. Defective mer receptor tyrosine kinase signalling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 1429–1431 (2008). References 90 and 91 provide in vivo evidence that the phagocyte receptor MERTK has an important role in mediating efferocytosis and thus preventing plaque necrosis in advanced mouse atheroma.
Aprahamian, T. et al. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J. Exp. Med. 199, 1121–1131 (2004).
Libby, P. et al. Macrophages and atherosclerotic plaque stability. Curr. Opin. Lipidol. 7, 330–335 (1996).
Li, Y. et al. Cholesterol-induced apoptotic macrophages elicit an inflammatory response in phagocytes, which is partially attenuated by the Mer receptor. J. Biol. Chem. 281, 6707–6717 (2006).
Cui, D. et al. Pivotal advance: macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J. Leukoc. Biol. 82, 1040–1050 (2007).
Sather, S. et al. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 109, 1026–1033 (2007).
Komura, H., Miksa, M., Wu, R., Goyert, S. M. & Wang, P. Milk fat globule epidermal growth factor-factor VIII is downregulated in sepsis via the lipopolysaccharide–CD14 pathway. J. Immunol. 182, 581–587 (2009).
Zou, W. et al. Caenorhabditis elegans myotubularin MTM-1 negatively regulates the engulfment of apoptotic cells. PLoS. Genet. 5, e1000679 (2009).
Xu, W. et al. IL-10-producing macrophages preferentially clear early apoptotic cells. Blood 107, 4930–4937 (2006).
Odegaard, J. I. et al. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab. 7, 496–507 (2008).
Mukundan, L. et al. PPARδ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nature Med. 15, 1266–1272 (2009).
Wilson, P. W., Castelli, W. P. & Kannel, W. B. Coronary risk prediction in adults (the Framingham Heart Study). Am. J. Cardiol. 59, 91G–94G (1987).
Aprahamian, T., Takemura, Y., Goukassian, D. & Walsh, K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin. Exp. Immunol. 152, 448–455 (2008).
Agrawal, A. et al. Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signalling pathway. J. Immunol. 178, 6912–6922 (2007).
Liew, F. Y., Xu, D., Brint, E. K. & O'Neill, L. A. Negative regulation of Toll-like receptor-mediated immune responses. Nature Rev. Immunol. 5, 446–458 (2005).
Foster, S. L., Hargreaves, D. C. & Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978 (2007).
Liu, J. et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nature Med. 15, 940–945 (2009).
Zernecke, A. et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 102, 209–217 (2008).
Saito, Y. et al. Effects of EPA on coronary artery disease in hypercholesterolemic patients with multiple risk factors: sub-analysis of primary prevention cases from the Japan EPA Lipid Intervention Study (JELIS). Atherosclerosis 200, 135–140 (2008).
Li, S. et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and its reversal by a fish oil diet. Circ. Res. 15 Oct 2009 (doi:10.1161/CIRCRESAHA.109.199570).
O'Sullivan, T. P. et al. Aromatic lipoxin A4 and lipoxin B4 analogues display potent biological activities. J. Med. Chem. 50, 5894–5902 (2007).
Burstein, S. H. & Zurier, R. B. Cannabinoids, endocannabinoids, and related analogues in inflammation. AAPS J. 11, 109–119 (2009)
Dol-Gleizes, F. et al. Rimonabant, a selective cannabinoid CB1 receptor antagonist, inhibits atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 29, 12–18 (2009).
Moreira, F. A. & Crippa, J. A. The psychiatric side-effects of rimonabant. Rev. Bras. Psiquiatr. 31, 145–153 (2009).
Maderna, P., Yona, S., Perretti, M. & Godson, C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac2–26 . J. Immunol. 174, 3727–3733 (2005).
Wickline, S. A., Neubauer, A. M., Winter, P. M., Caruthers, S. D. & Lanza, G. M. Molecular imaging and therapy of atherosclerosis with targeted nanoparticles. J. Magn. Reson. Imaging 25, 667–680 (2007).
Chen, Y. et al. Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J. Immunol. 183, 1346–1359 (2009).
Hallett, J. M. et al. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol. Sci. 29, 250–257 (2008).
Rouleau, J. Improved outcome after acute coronary syndromes with an intensive versus standard lipid-lowering regimen: results from the Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) trial. Am. J. Med. 118, S28–S35 (2005).
Strong, J. P., Malcom, G. T., Oalmann, M. C. & Wissler, R. W. The PDAY study: natural history, risk factors, and pathobiology. Pathobiological Determinants of Atherosclerosis in Youth. Ann. NY Acad. Sci. 811, 226–237 (1997).
Acknowledgements
The author acknowledges stimulating conversations with E. A. Fisher, E. Thorp, D. Schrijvers, G. Randolph, L. Chan and A. Chawla during the conception and writing of this Review. The work from the author's laboratory cited here was supported by grants HL54591 and HL75662 from the US National Heart, Lung and Blood Institute.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Related links
Glossary
- Efferocytosis
-
The phagocytic clearance of apoptotic cells (from the Latin 'effero', meaning to take to the grave or bury) before they undergo secondary necrosis. The process usually triggers an anti-inflammatory response.
- Alternatively activated macrophage
-
(M2 macrophage). A macrophage stimulated by IL-4 or IL-13 that expresses arginase 1, the mannose receptor CD206 and IL-4 receptor-α. There may be pathogen-associated molecular patterns expressed by helminths that can also drive the alternative activation of macrophages.
- Atherosclerosis
-
A process whereby lipids, inflammatory cells and extracellular matrix accumulate in the subendothelial space (intima) of focal areas of medium-sized arteries, which finally leads to plaque formation.
- Atherothrombotic vascular disease
-
(ATVD). Disease caused by acute occlusive arterial thrombosis overlying areas of chronic atherosclerosis. The occlusive thrombosis starves the tissue that the involved artery feeds of oxygen and nutrients. For example, if the involved artery feeds the heart muscle, myocardial infarction (death of heart muscle cells) can ensue.
- Insulin resistance
-
A state in which signalling through insulin receptors is impaired. The cause can be exposure to high levels of insulin, which downregulates insulin receptors by a negative homeostatic mechanism, or disruption of signalling molecules downstream of the insulin receptor, such as insulin receptor substrate 1 (IRS1) and IRS2. Insulin resistance, caused by high levels of insulin in the bloodstream, is responsible for a substantial portion of the pathology associated with type 2 diabetes, including ATVD.
- Atherosclerotic plaque
-
The name given to an atherosclerotic lesion without precise designation of lesion stage, but usually referring to a lesion that has developed beyond the early foam cell stage, particularly a lesion that is raised and fibrotic.
- Atherothrombosis
-
Following the rupture of unstable atherosclerotic plaques, thrombogenic material becomes exposed or released to mediate thrombus formation and eventually occlusion of an artery.
- Secondary necrosis
-
A process that occurs in apoptotic cells that are not cleared by phagocytes. The integrity of the plasma membrane is lost and the constituents of the cell are released.
- Atheroma
-
An advanced atherosclerotic plaque, particularly one that is rich in cholesterol-filled macrophage foam cells and has areas of plaque necrosis.
- Apolipoprotein E (Apoe)−/− mice
-
A widely used mouse model that is prone to develop atherosclerosis because the mice have high levels of types of atherogenic lipoprotein called remnant lipoproteins. This lipoprotein abnormality is cause by the genetic absence of apolipoprotein E (APOE), which normally clears remnant lipoproteins from the bloodstream by interacting with hepatocytes.
- Endoplasmic reticulum (ER) stress
-
Perturbation of ER function, such as that which occurs during a high level of protein translation or when newly synthesized proteins become misfolded, resulting in the activation of a corrective signal transduction pathway called the unfolded protein response.
- Low-density lipoprotein receptor (Ldlr)−/− mice
-
Another widely used mouse model of atherosclerosis. These mice accumulate high levels of low-density lipoprotein (LDL) when on a high-fat diet because their hepatocytes lack LDL receptors and cannot efficiently rid the bloodstream of atherogenic LDL particles.
- Atherosclerotic lesion
-
A collection of lipids, cells and extracellular matrix in a focal area of the arterial subendothelium. These lesions are triggered by the accumulation of apolipoprotein B-containing lipoproteins and a maladaptive, macrophage-dominant inflammatory response to these lipoproteins. With further intracellular lipid accumulation and formation of foam cells, atherosclerotic plaques develop. The later-stage lesions contain a core of extracellular lipid surrounding the cholesterol-laden cells, many of which undergo apoptosis or necrosis.
- Periodontal disease
-
A bacteria-mediated inflammatory disease of the gums that has an epidemiological association with atherothrombotic vascular disease, perhaps through promoting systemic inflammation.
- Unfolded protein response
-
(UPR). A response that increases the ability of the endoplasmic reticulum (ER) to fold and translocate proteins, decreases the synthesis of proteins degrades misfolded proteins and corrects disturbances in calcium and redox imbalance in the ER. If prolonged, the UPR can trigger apoptosis.
- Foam cell
-
A macrophage in the arterial wall that ingests cholesterol-rich apolipoprotein B-containing lipoproteins and thereby accumulates cholesteryl fatty acid esters. These cells secrete various substances involved in plaque growth.
- Statins
-
A family of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), an enzyme that catalyses the conversion of HMG-CoA to L-mevalonate. These molecules are mainly used as cholesterol-lowering drugs, but they also have immunoregulatory and anti-inflammatory properties, the clinical significance of which is not fully known. L-mevalonate and its metabolites are implicated in cholesterol synthesis and other intracellular pathways.
Rights and permissions
About this article
Cite this article
Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 10, 36–46 (2010). https://doi.org/10.1038/nri2675
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri2675
This article is cited by
-
Macrophage profiling in atherosclerosis: understanding the unstable plaque
Basic Research in Cardiology (2024)
-
Genetic dissection of the impact of lncRNA AI662270 during the development of atherosclerosis
Journal of Translational Medicine (2023)
-
Macrophage Anti-inflammatory Behaviour in a Multiphase Model of Atherosclerotic Plaque Development
Bulletin of Mathematical Biology (2023)
-
A Lipid-Structured Model of Atherosclerotic Plaque Macrophages with Lipid-Dependent Kinetics
Bulletin of Mathematical Biology (2023)
-
Performance of novel 3D printing tools in removing coronary-artery calcification tissue
Bio-Design and Manufacturing (2023)
