
Abstract
Healing of the inflamed mucosa (mucosal healing) is an emerging new goal for therapy and predicts clinical remission and resection-free survival in inflammatory bowel diseases (IBDs). The era of antitumor necrosis factor (TNF) antibody therapy was a remarkable progress in IBD therapy and anti-TNF agents led to mucosal healing in a subgroup of IBD patients; however, many patients do not respond to anti-TNF treatment highlighting the relevance of finding new targets for therapy of IBD. In particular, current studies are addressing the role of other anticytokine agents including antibodies against interleukin (IL)-6R, IL-13, and IL-12/IL-23 as well as new anti-inflammatory concepts (regulatory T cell therapy, Smad7 antisense, Jak inhibition, Toll-like receptor 9 stimulation, worm eggs). In addition, blockade of T-cell homing via the integrins α4β7 and the addressin mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) emerges as a promising new approach for IBD therapy. Here, new approaches for achieving mucosal healing are discussed as well as their implications for future therapy of IBD.
Similar content being viewed by others

Introduction
Inflammatory bowel diseases (IBD; Crohn’s disease (CD), ulcerative colitis (UC)) are chronic relapsing diseases that lead to structural damage with destruction of the bowel wall.1, 2, 3 Clinically, IBD patients can suffer from chronic diarrhea, malabsorption, weight loss, rectal bleeding, abdominal pain, stenoses, abscesses, and fistula formation, and many patients require surgery over time.4 In addition, various extraintestinal manifestations (primary sclerosing cholangitis, ankylosing spondylitis, iritis/uveitis, pyoderma gangrenosum, erythema nodosum) and immune-mediated diseases (asthma, psoriasis, rheumatoid arthritis) are associated with IBD.4 Finally, there is an increased risk of colitis-associated neoplasias in IBD, particularly in patients with ulcerative pancolitis and colonic CD.5, 6 Given the clinical relevance of these IBD-related problems, it is no surprise that IBD has a major impact on quality of life including fatigue, relationships, and employment.7
Although clinical IBD studies in the past have mainly focused on improvement of diarrhea and abdominal pain, mucosal healing on endoscopy has recently emerged as a key treatment goal in IBD.8, 9 Although there is still no general consensus on the definition of the term mucosal healing, mucosal healing on endoscopy usually refers to resolution of ulcers in CD and erosions and ulcers in UC.10, 11 Interestingly, mucosal healing in IBD was associated with more effective disease control, more frequent steroid-free remission of disease, lower rates of hospitalization and surgery, and improved quality of live as compared with conventional treatment goals.12 These findings highlight the role of mucosal healing for therapy of IBD. In spite of the fact that not all groups agree on the relevance of mucosal healing in the absence of large prospective intervention studies,13 mucosal healing has become a key end point in clinical trials and predicts sustained clinical remission and resection-free survival of IBD patients.8, 12, 14 New treatment options thus will have to demonstrate their potential to induce mucosal healing in IBD.
The structural basis of mucosal healing
Various studies in recent years have highlighted a crucial role of molecular signaling events for mucosal healing in IBD. It was found that mucosal healing relies on the coordinated activity of intestinal epithelial cells (IECs), goblet cells, and Paneth cells for improvement of intestinal barrier function.15, 16, 17
After surface injury, IECs surrounding the wounded area lose their columnar polarity and quickly migrate to the lesion to initiate wound healing. This process is called “epithelial restitution,” which begins within minutes after injury and has been identified as a crucial step in resealing of the injured surface.17 Epithelial restitution was found to be independent of cell proliferation and regulated by cytokines. Tumor growth factor (TGF)-α, epidermal growth factor, interleukin (IL)-1β, and interferon (IFN)-γ augmented epithelial restitution in wounded IEC monolayers by inducing the production of bioactive TGF-β1 in epithelial cells.18 However, TGF-β-independent pathways to induce epithelial restitution exist. In fact, it has been shown that secreted trefoil peptides, galectin-2 and galectin-4, promote IEC restitution independent of TGF-β.16, 19 Furthermore, changes of the actin cytoskeleton induced by small GTPases of the Rho family are required for epithelial reconstitution20 and this process is modulated by chemokines including CXCR4 and CXCL12.21 In addition, CCL20 and human β-defensin 2 have been shown to favor epithelial restitution by inducing cell migration of IECs without inducing cell proliferation.22
Epithelial restitution is followed by additional steps in wound healing such as increased epithelial cell proliferation and differentiation. These processes start after epithelial restitution within hours or days after injury.23, 24 Cell proliferation is important to increase the pool of IECs available to resurface the defect, but requires tight control mechanisms to prevent uncontrolled proliferation. Accordingly, IECs receive signals via growth factors such as epidermal growth factor, cytokines such as IL-6 and IL-22, and Toll-like receptor (TLR) ligands such as lipopolysaccharide (LPS) and CplusG DNA to induce activation of master transcription factors (e.g. nuclear factor-κB, signal transducer and activator of transcription 3 (STAT3)), cell survival, and cell proliferation.8, 25, 26 For instance, both IL-6 and IL-22 are known to boost proliferation of IECs by activation of the transcription factor STAT3.27, 28 The relevance of such regulation was highlighted by the finding that inactivation of STAT3 in IECs suppresses experimental wound healing in vivo. In contrast to IL-6 and IL-22, the proinflammatory cytokines IFN-γ and tumor necrosis factor (TNF) were found to block IEC proliferation and impaired wound healing by favoring IEC apoptosis.29, 30 However, growth hormone was shown to induce activation of epithelial STAT5 and support wound healing. Consistently, STAT5 knockout in IECs led to impaired experimental wound healing by activation of epithelial myosin light-chain kinase with subsequent alterations of tight junctions and barrier function.31, 32 In addition to cytokines, prostaglandin E2 contributes to survival of IECs in tissue injury and prevents IEC apoptosis. Importantly, regulatory factors for epithelial proliferation are not only derived from IECs themselves but also from immune and nonimmune cells in the local microenvironment of the gut. Although macrophages and T cells are known to produce IL-6 and TNF to regulate IEC proliferation and survival, fibroblasts were found to produce hepatocyte growth factor to induce IEC proliferation.28, 33, 34 Furthermore, granulocyte–macrophage colony-stimulating factor-induced dendritic cells were found to regulate wound healing of IECs.35
TLRs are additional key regulator of wound healing.36 In the healthy mucosa, TLRs act as innate receptors for pathogen-associated molecular patterns in commensal bacteria. The involvement of specific TLRs in mucosal wound healing has been documented by numerous studies in recent years. TLR2 has been shown to selectively induce synthesis of trefoil factor TFF3, thereby favoring survival of IECs and mucosal wound healing.37 In these studies, TLR2-deficient mice demonstrated increased morbidity and mortality during acute colonic injury and this phenotype could be rescued by administration of recombinant TFF3.38 Additional studies indicated a regulatory role of the LPS ligand TLR4 in wound healing.39 In acute mucosal injury during dextran sulfate sodium colitis, TLR4−/− deficiency was associated with reduced infiltration of inflammatory cells compared with wild-type mice. TLR4-deficient mice showed impaired neutrophil recruitment with diminished macrophage inflammatory protein-2 expression by lamina propria macrophages. Furthermore, TLR4 was found to limit bacterial translocation during mucosal injury, suggesting that TLR4 controls mucosal wound healing through several mechanisms. Finally, recent studies suggested that TLR9 has a crucial role in mucosal wound healing. TLR9 serves as a ligand for CplusG motifs in bacterial DNA and thus acts as a link between the commensal microflora and the innate immune system in the gut. TLR9 expression was found on the apical and basolateral membranes in IECs.40, 41 However, although apical TLR9 stimulation induced intracellular tolerance to subsequent TLR challenges and restricted inflammation, basolateral TLR9 activation resulted in proinflammatory nuclear factor-κB responses. TLR9-deficient mice were found to be more susceptible to acute dextran sulfate sodium injury and demonstrated delayed wound repair highlighting the protective role of TLR9 in wound healing. Mechanistically, TLR9 deficiency caused reduced gene expression of hairy enhancer of split 1, an epithelial differentiation factor, and vascular endothelial growth factor, a growth factor controlling epithelial cell restitution.42 The potential therapeutic relevance of these observations was highlighted by the finding that a TLR9 agonist ameliorated mucosal injury and inflammation by inducing type I interferons.43 Moreover, immunostimulatory DNA and probiotics appear to be mediated by their protective effects on barrier function, mucosal healing, and intestinal inflammation by activation of TLR9 signaling.44, 45 Thus, in contrast to other TLR ligands such as LPS that may induce overwhelming immune responses upon administration, TLR9 ligands selectively induced anti-inflammatory immune responses and strengthened barrier function.
Cell proliferation, expansion, migration, and differentiation ultimately lead to closure of erosions and ulcerations. This process is supported by antimicrobial peptides and products released by Paneth cells such as defensins and REG proteins (regenerating protein family) that have an important role in intestinal homeostasis3, 46 as well as by mucins produced by goblet cells that prevent translocation of commensal bacteria with subsequent immune cell activation. Recently, the cysteine-rich domains of the intestinal mucin mucin-3 (MUC3) were found to promote IEC migration, inhibit IEC apoptosis, and accelerate experimental wound healing.47 In some cell lines, human β-defensin 2 augmented mucin production (MUC2, MUC3) and prevented TNF-mediated IEC apoptosis, suggesting that defensins have pleiotropic protective functions in experimental wound healing in addition to their effects on epithelial restitution.48
Given the clinical relevance of mucosal healing for patients with IBD, it is of major relevance to identify new targets for therapy that may induce mucosal healing. Thus, in the following paragraphs, we will discuss novel approaches for therapy of IBD and their potential impact on mucosal inflammation.
Modulation of microbiota, barrier function, and innate immunity in IBD
The commensal microflora in healthy humans is estimated at 1013 to 1014 with a complex microbial community.49 Given the high numbers of antigen-presenting cells and lymphocytes in the normal intestine, it is clear that an intact barrier function is essential for prevention of uncontrolled mucosal inflammation.26 The gut barrier comprises IECs with tight junctions, adherens junctions, and desmosomes as well as antimicrobial peptides and mucins.17, 50
Numerous studies in recent years have described alterations in barrier function in IBD that favor diarrhea and uptake of potentially noxious antigens50, 51 (Figure 1). For instance, alterations of IECs in IBD comprise reduced numbers of horizontal tight junction strands and altered tight junction protein expression. Furthermore, IECs in IBD have been described to be more susceptible to undergo apoptosis, leading to structural defects of the bowel wall with erosions and ulcerations. The structural changes of IECs in IBD are likely due to proinflammatory cytokines such as TNF, IFNγ, and IL-13, which are upregulated in the inflamed mucosa (Figures 2 and 3).50, 52 These cytokines favor IEC apoptosis and may thus have an important role in the altered barrier function in IBD. For instance, TNF favors alterations of tight junctions and cell death of IEC via TNFR1 signaling, indicating that this cytokine induces barrier alterations in IBD.53 Indeed, anti-TNF antibody therapy has been shown to improve IEC-controlled barrier function in IBD patients, suggesting that this treatment may have a profound effect on IEC function in vivo.54 Furthermore, IL-13 is highly produced by T cells in patients with UC and impairs barrier function.55 Thus, antibodies against IL-13 might be a new approach for UC therapy and clinical studies addressing this concept are currently ongoing (Figure 4).

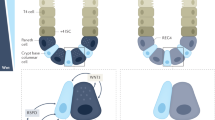
Signaling pathways controlling wound healing and intestinal inflammation. Intestinal epithelial cells, goblet cells, and Paneth cells receive signals from the local microenvironment of the gut. In particular, cytokines, Toll-like receptor (TLR) ligands and growth factors induce cellular signaling events, leading to apoptosis or proliferation and cell expansion, respectively. Defects in goblet cells may result in impaired mucin production, whereas alterations of Paneth cells cause reduced production of antimicrobial peptides such as defensins. AIEC, adherent invasive Escherichia coli; GH, growth hormone; LPS, lipopolysaccharide; EGF, epidermal growth factor; FGF, fibroblast growth factor; GH, growth hormone; IL, interleukin; IFN, interferon; NF-κB, nuclear factor-κB; PGE2, prostaglandin E2; REG, regenerating protein family; STAT, signal transducer and activator of transcription.

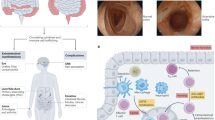
Hypothetical pathogenesis of inflammatory bowel disease (IBD). Although ulcerative colitis (UC) affects the mucosa and submucosa, inflammation may affect all layers of the bowel wall in Crohn’s disease (CD). Pathogenesis of IBD is related to an altered barrier function in a genetically susceptible host allowing translocation of bacteria and bacterial antigens into the mucosa. Antigens are taken up by antigen-presenting cells such as dendritic cells and macrophages, followed by antigen presentation to T cells. T lymphocytes enter into the lamina propria via homing from the blood stream and undergo T helper cell polarization upon antigen presentation. Although T helper type 1 (Th1) T cells expressing T-bet predominate in CD,121, 140, 193 T cells producing interleukin (IL)-5 and IL-13 are found in UC. Th17 cells expressing RAR-related orphan receptor A/C (RORA/C) have been described in UC and CD.124, 194 Finally, a cell population expressing both Th17 and regulatory T cell (Treg) markers has been recently been described in CD.129 MadCAM-1, mucosal vascular addressin cell adhesion molecule 1; TNF, tumor necrosis factor.

Signaling events in inflammatory bowel diseases. (a) Cytokine signaling events that have major effects of T-cell activation and survival in inflammatory bowel disease (IBD) patients. Although the interleukin (IL)-12/IL-23 pathway is relevant for Crohn’s disease (CD) pathogenesis, the IL-6/soluble IL-6R (sIL-6R) pathway is additionally being explored for IBD therapy. Finally, the membrane-bound tumor necrosis factor (mTNF)/tumor necrosis factor receptor 2 (TNFR2) signaling pathway is important for the pathogenesis of both CD and ulcerative colitis (UC). (b) The IL-6 signaling pathway in IBD. Membrane-bound IL-6R (mIL-6R) undergoes shedding via proteases such as TACE (tumor necrosis factor-converting enzyme) and ADAM17 (ADAM metallopeptidase domain 17), resulting in the production of the sIL-6R.134 IL-6 together with the sIL-6R activates the gp130 target molecule on both T cells and epithelial cells. This results in signal transducer and activator of transcription 3 (STAT3) activation and cell activation and survival. In this way, IL-6 signaling favors T-cell resistance against apoptosis in IBD. In addition, such signaling favors growth of epithelial neoplasias in IBD (modified according to refs. 28,131,132). (c) Potential mechanism of action of anti-TNF antibodies in IBD. Anti-TNF antibodies block the interaction between mTNF on CD14+ antigen-presenting cells with TNFR2 on lamina propria T cells. Accordingly, this treatment suppresses TNF receptor-associated factor 2 (TRAF2) and nuclear factor-κB (NF-κB) activation with IL-6 production and T-cell survival. Subsequently, anti-TNF antibodies induce programmed cell death (apoptosis) of T cells in the inflamed gut and such apoptosis correlated to the clinical efficacy of anti-TNF therapy (modified according to ref. 112). EGFR, epidermal growth factor receptor.

New therapeutic concepts in inflammatory bowel diseases. (a and b) New therapeutic strategies in Crohn’s disease (CD) and ulcerative colitis (UC) that are currently tested in phase 1 to phase 3 trials. Targets, drugs, and antibodies are shown for both diseases. Clinically approved drugs are indicated at the bottom of both panels (Natalizumab is not approved for CD therapy in Europe). Red dots indicate cytokine targets, whereas blue dots represent other targets. One dot is on the circle because it represent a phase 1/2 study rather than a pure phase 1 or phase 2 study. (c) Cytokine receptors targeted by the Jak1/Jak3 inhibitor tofacitinib in inflammatory bowel disease. This Jak inhibitor has shown promising results in patients with UC and is currently under further clinical development. A modified schematic diagram of cytokine signaling via Jak proteins according to Liao et al.159 is shown. IL, interleukin; MadCAM-1, mucosal vascular addressin cell adhesion molecule 1; STAT, signal transducer and activator of transcription; TGF, tumor growth factor; TNF, tumor necrosis factor.
In addition to IECs, changes of barrier function in IBD comprise impaired mucin production.56, 57 This problem is particularly relevant for the intestinal barrier, as mucins prevent a direct contact of the commensal microflora with IECs. In the normal human intestine, the mucus is organized around glycosylated MUC2 produced by goblet cells. It has been suggested that O-glycans on MUC2 may serve as nutrients for bacteria and have a role in the selection of the local bacterial strains. However, there are important regional differences with regard to the structure of the mucus barrier. In the murine large intestine, two mucus layers have been identified. Although the inner, dense mucus layer is devoid of bacteria and firmly adherent to IECs, the outer nonattached mucus layer contains commensal bacteria.58, 59 In contrast to the large bowel, the mucus layer in the small intestine is discontinuous and thinner.
Severe alterations of the inner mucosal mucus layer have been noted in IBD, particularly in the colon of patients with UC. In these patients, expression levels of secretory MUC2 and transmembrane MUC3 and MUC4 proteins were found to be low or absent.60 Thus, it is potentially interesting for therapy of IBD to overcome the impaired barrier function by modulation of mucin function or expression. One approach to circumvent the loss of mucin-controlled barrier function in UC that has been clinically tested consists of phosphatidylcholine (lecithin).61 Whereas the mucus on the surface of the large intestine normally contains high concentrations of phosphatidylcholine in lamellar structures, phosphatidylcholine is substantially reduced in UC but not CD patients and this reduction may favor bacterial translocation. This finding has led to the concept to treat patients with phosphatidylcholine.62 Indeed, in controlled clinical studies, delayed release phosphatidylcholine given daily was significantly superior to placebo in induction of remission in both patients with non-steroid-treated active UC and steroid-dependent active UC. Future phase 3 studies will have to confirm these observations to underline the relevance of the concept that phosphatidylcholine strengthens barrier function and prevents translocation of commensal bacteria with subsequent immune cell activation in UC.
Various studies in recent years have attempted to suppress chronic intestinal inflammation by inducing anti-inflammatory pathways via antimicrobial peptides. One example consists of the induction of defensins as antimicrobial peptides. Defensins are produced by Paneth cells in the terminal ileum and these cells are markedly reduced in active CD because of TNF-mediated necroptosis.63 Furthermore, reduced expression of the α-defensins HD5 and HD6 by Paneth cells has been found in ileal CD, whereas production of the β-defensin HBD1 by goblet cells was suppressed in colonic CD.46 These studies have raised expectations that modulation of defensin levels might be used for treatment of gut inflammation in CD, particularly for maintenance of remission. Although treatment with recombinant defensins has not been tested in clinical trials, it was found that overexpression of HD5 in transgenic mice results in resistance against infection with orally administered Salmonella typhimurium and experimental colitis.64 This finding is at least consistent with the idea that defensins might be used for IBD therapy. Furthermore, the probiotic strain Escherichia coli Nissle has been shown to induce defensin production and was as effective as 5-aminosalicylates in maintaining remission in patients with UC.65, 66 It is thus possible that induction of defensin production contributes to the clinical efficacy of E. coli Nissle in UC. However, in contrast to this disease, probiotic strains such as E. coli Nissle have not been very effective in CD so far.
As bacteria use CplusG sequence motifs to induce immune cell activation via TLR9 signaling, modulation of such innate immune responses is another new approach for IBD therapy. As CpG DNA has been successfully used for therapy of experimental colitis,45, 67 this concept has been recently tested in a small study for steroid-dependent or steroid-refractory UC patients by topical administration of a CpG oligonucleotide (DIMS0150).68 However, future studies will have to unequivocally prove the efficacy of this treatment in UC.
In addition to probiotics, antibiotics have been used for modulation of the commensal microflora in IBD. In contrast to other antibiotics, pilot studies suggested that oral administration of the antibiotic agent rifaximine induces remission in CD69, 70 and that metronidazole and ornidazole suppress postoperative relapses in this disease.71 However, large phase 3 studies are missing and antibiotics are therefore not yet standard of care for IBD therapy in the absence of systemic infections.
Recent evidence suggests that IBD patients may be exposed to uncontrolled immune cell activation upon translocation of bacteria because of the defects in autophagy.72 In fact, single-nucleotide polymorphisms in ATG16L1 and IRGM genes have been identified in CD patients and both genes have been shown to have a crucial role in autophagy/xenophagy during innate immune responses.73 For instance, ATG16L1 and IRGM both control antibacterial autophagy of Salmonella typhimurium and adherent invasive E. coli (AIEC). This finding is of potential interest, as AIECs are thought to have an important regulatory role in CD.74 Additional CD susceptibility genes such as nucleotide oligomerization domain 2 may also control autophagy/xenophagy in the inflamed intestine.75 As defects in xenophagy may cause impaired clearance of bacteria and inappropriate immune responses, it is likely that these findings contribute to changes in innate immunity in IBD. Prevention of inappropriate handling of translocated bacteria may thus be relevant for IBD therapy. In fact, AIECs present in inflamed CD mucosa in the terminal ileum require TNF to favor their replication in infected lamina propria macrophages.76 Anti-TNF treatment has been shown to suppress AIEC replication in infected immune cells and this observation might open new avenues for therapy in the future.
Taken together, substantial progress has been made in our understanding of the coordinated interplay between the commensal microflora, antimicrobial peptides, and innate immunity in IBD. It has become increasingly clear that loss of barrier function is a key phenomenon in IBD that favors secretory diarrhea and translocation of potentially noxious bacterial antigens.50, 51 The latter point is of particular relevance, as translocation of commensal bacteria into the mucosa is followed by expansion of antigen-presenting cells and T cells that produce proinflammatory cytokines to induce and maintain chronic intestinal inflammation. However, with the important exception of probiotic therapy with E. coli Nissle, these approaches have not reached clinical routine as yet. Future studies will have to demonstrate the relevance of modulation of the microflora for IBD therapy.
Modulation of adaptive immunity in IBD: classical immunosuppressive drugs
Most established therapies for IBD have major effects on the adaptive immune system. For instance, corticosteroids that are used for induction of clinical remission in both CD and UC strongly suppress proinflammatory cytokine production by macrophages and T cells.77 Furthermore, cyclosporin A and tacrolimus that are effective for induction of remission in severely active UC78, 79 and methotrexate that is used for chronically active CD80, 81 are known to suppress proinflammatory cytokine production by T cells.82, 83, 84, 85 The former drugs are known to bind to cyclophilins in T lymphocytes to interfere with Ca signaling and inhibit activity of NFAT (nuclear factor of activated T cells) transcription factors (NFATc1–4). NFATc2 in turn has been shown to control IL-6 production and T-cell survival in colitis.86, 87 Finally, azathioprine and 6-mercaptopurine are effective in maintenance of remission in both CD and UC by blocking T-cell activation.88, 89 Specifically, it was found that these thiopurine drugs block T-cell costimulation by interfering with the vav1/Rac1 signaling pathway in T cells. Azathioprine- and 6-mercaptopurine-derived 6-thio-GTP has been shown to bind to the small GTPase Rac1 to block guanosine exchange activity of vav1 on this protein. Consecutively, Rac1 activity is blocked over time, leading to inhibition of ERM (ezrin–radixin–moesin)-dependent immunological synapse formation between antigen-presenting cells and T cells. Furthermore, suppression of Rac1 activation induces T-cell apoptosis (via blockade of STAT3 activation). The clinical relevance of this observation was highlighted by the finding that clinical responsiveness to thiopurine therapy correlates to the induction of T-cell apoptosis in the inflamed intestine in IBD.88 Collectively, the above findings indicate that classical immunosuppressive drugs that are successfully used for IBD therapy specifically interfere with the adaptive immune system. As not all IBD patients respond to this type of therapy and as these drugs may cause severe side effects in some patients, it is of high clinical relevance to define novel drugs that block specific signaling pathways in cells of the adaptive immune system. In the following part of the review, the development of anti-TNF antibody therapies for IBD as well as some novel approaches to attack key proinflammatory pathways in chronic intestinal inflammation are discussed.
Attacking key proinflammatory pathways in IBD: anti-TNF antibodies and beyond
Macrophages and T lymphocytes produce large amounts of the proinflammatory cytokine TNF-α in IBD and this cytokine has a pivotal role in IBD pathogenesis.2 These cells produce TNF as a transmembrane protein (mTNF) whose soluble form (sTNF) is released by proteolytic cleavage via the protease TACE (tumor necrosis factor-converting enzyme)/ADAM17 (ADAM metallopeptidase domain 17).90 Thus, two forms of TNF exist in the inflamed gut and these forms appear to exert different functions in IBD. Although sTNF is released by immune cells to deliver signals in the local environment or systemic circulation by binding to TNF-R1 on target cells, mTNF is mainly expressed on the surface of CD14+ mucosal macrophages, and preferentially binds to TNF-R2 on lamina propria T cells (Figure 3).91
The clinical development of neutralizing antibodies against TNF has been a crucial milestone for IBD therapy. In fact, randomized controlled trials found that the anti-TNF antibodies adalimumab, certolizumab pegol, and infliximab were effective for induction and maintenance of remission in both CD and UC.92, 93, 94, 95, 96 Furthermore, anti-TNF therapy was found to induce fistula closure in CD97 and induced mucosal healing in both CD and UC.10, 98 On the basis on these landmark studies, anti-TNF therapy has been approved for treatment of patients with moderate-to-severe IBD. Although infliximab is a chimeric anti-TNF antibody with murine sequence parts (intravenous administration), certolizumab is a humanized, pegylated anti-TNF Fab fragment and adalimumab is a fully human anti-TNF antibody (subcutaneous administration). In addition to these antibodies, there are numerous new developments in the anti-TNF field. This includes the fully human anti-TNF antibody golimumab that has recently successfully passed phase 3 studies in UC (subcutaneous administration) (Figure 4).99, 100 Furthermore, small anti-TNF molecules given orally, biosimilars, and nanobodies are currently being developed for clinical use.101 In this context, one interesting concept consists of genetically modified bacteria-producing anti-TNF nanobodies.102 Daily oral administration of nanobody-secreting Lactobacillus lactis resulted in local delivery of anti-TNF nanobodies in the gut and led to significantly reduced inflammation in mice with dextran sulfate sodium-induced experimental chronic colitis. Finally, active immunization with a therapeutic vaccine against TNF is another alternative as anti-TNF targeting strategy that is potentially devoid of some treatment limitations of current anti-TNF-blocking agents.103 Indeed, active immunization with the so-called anti-TNF kinoid has recently been tested in patients with CD and induced polyclonal anti-TNF antibodies in these patients. Future clinical trials will determine the relevance of this strategy for anti-TNF therapy in IBD patients.
In spite of the clinical efficacy of anti-TNF antibody treatment in IBD, however, a subgroup of patients do not respond to anti-TNF therapy within 4–12 weeks after initiation of therapy.104, 105, 106 These patients show little or no changes of clinical symptoms and no mucosal healing upon anti-TNF therapy but are potentially exposed to side effects of this type of therapy, such as infections, reactivation of tuberculosis, allergic reactions, skin disorders, demyelinating disorders, and lupus-like autoimmunity.107 Potential reasons for failure of anti-TNF antibody therapy include the development of inhibitory antibodies (human antichimera or human antihuman antibodies) as well as insufficient antibody trough levels in the serum due to underdosing.108 Furthermore, there is the possibility of mechanistic failure of antibody therapy caused by TNF-independent gut inflammation. It is thus of interest to understand the mechanism of action of anti-TNF antibodies in IBD to identify reasons for therapy failure.
Although several potential mechanisms of action have been proposed for anti-TNF therapy in IBD (e.g. barrier improvement, direct cytotoxicity, induction of regulatory macrophages, and T cells),54, 109, 110 several lines of evidence suggest that the induction of T-cell apoptosis is a key mechanism of action of anti-TNF antibodies. First, in an elegant series of studies involving in vivo imaging with radiolabeled annexin V antibodies, it was found that T-cell apoptosis occurs within the first days after the initiation of anti-TNF therapy in IBD.111 Moreover, the induction of apoptosis was directly correlated to the rapid clinical response of the patients and patients without the induction of T-cell apoptosis showed little or no response to anti-TNF therapy. Additional studies revealed that clinically effective anti-TNF antibodies do not directly induce T-cell apoptosis of purified primary lamina propria CD4+ T cells in IBD patients, although these antibodies are capable of inducing T-cell death directly in tumor cell lines via induction of bax and bak.112, 113, 114 Instead, it was found that the anti-TNF antibodies infliximab, adalimumab, and certolizumab pegol bind to mTNF, block the interaction between mTNF on CD14+ macrophages and TNFR2 on lamina propria T cells, and suppress TNF receptor-associated factor 2 and nuclear factor-κB activation in T cells followed by T-cell apoptosis (Figure 3). Thus, it appears that the clinically effective anti-TNF antibodies adalimumab, certolizumab pegol, and infliximab mainly work by blocking mTNF signaling to induce T-cell death. This concept is supported by the clinical observation that the fusion protein etanercept that mainly blocks sTNF rather than mTNF signaling was not effective for the therapy of IBD.115 Furthermore, there is recent direct evidence that expression of mTNF on mucosal target cells is important for clinical efficacy of anti-TNF antibodies. In a phase 1 study of 25 in patients with active CD, mTNF expression on gut immune cells was visualized by topical administration of a fluorescent anti-TNF antibody (adalimumab), followed by confocal laser endomicroscopy for mTNF imaging. Interestingly, subsequent clinical therapy with adalimumab revealed that patients with high amounts of mTNF+ cells in the mucosa were significantly more likely to respond to anti-TNF therapy than patients with low amounts of mTNF+ cells.116 This observation opens new avenues for individualized adalimumab therapy in IBD and suggests that failure to anti-TNF therapy may occur in patients with low amounts of mTNF+ lamina propria cells due to TNF-independent gut inflammation. If this concept holds true, it might be of interest to target the mTNF/TNFR2 interaction specifically in the future rather than total TNF (mTNF plus sTNF). This approach might also avoid some potential side effects of anti-TNF therapy such as the reactivation of tuberculosis that is likely related to the blockade of the sTNF/TNFR1 interaction. In any case, the key role of the mTNF/TNFR2 pathway has also been highlighted in animal models of IBD117 and is an important difference to other immune-mediated diseases such as rheumatoid arthritis, where sTNF appears to be very important for disease pathogenesis and where etanercept is clinically highly effective.118
The findings that anti-TNF therapy is not effective in a relevant subgroup of IBD patients and that response to this therapy is lost in an additional subgroup of patients (up to 60% during the first year in CD patients) have led to an intensive search for novel therapeutic approaches. One concept derived from these studies was that combination therapy of the anti-TNF antibody infliximab plus azathioprine was significantly better than monotherapy with infliximab or azathioprine in both CD and UC.92 Thus, a combined top-down therapy with these agents might be useful in patients with severely active disease to prevent further complications. Furthermore, additional neutralizing antibodies against proinflammatory cytokines, chemokines, or costimulatory pathways have been clinically tested in IBD to identify new targets beyond TNF. In the following paragraphs, some of the recent new clinical and experimental approaches for therapy of chronic intestinal inflammation are reviewed.
Targeting new cytokines, chemokines, and costimulatory pathways in IBD
The exciting results of the anti-TNF trials have raised expectations that selective targeting of other cytokines in IBD might be as good or even better than targeting of TNF. Indeed, targeting of other cytokines beyond TNF has been effective in other diseases such as rheumatoid arthritis and psoriasis.119, 120 As CD is associated with a mixed T-helper type 1 (Th1) and Th17 cytokine profile,121, 122, 123, 124 targeting of cytokines from these T-cell subsets appeared to be of potential relevance for CD therapy (Figures 1 and 3). Unfortunately, anti-IFN-γ therapy with the humanized antibody fontolizumab did not result in major improvement of CD symptoms in moderate-to-severe disease activity, although a decrease of CRP levels was noted.125, 126 This led to the hope that targeting of Th17 cytokines might be more effective than targeting IFN-γ. However, although RORγ-expressing Th17 cells producing IL-17A/F have been shown to control experimental colitis in mice,127 targeting of IL-17A alone (via the monoclonal antibody secukinumab) did not improve but rather aggravated clinical symptoms in CD.128 Similarly, anti-IL-17R antibody therapy did not show clinical efficacy in CD, suggesting that targeting of IL-17A or IL-17R signaling in not suitable for CD therapy. The reasons for these disappointing results are not clear. It is possible that targeting of Th17 cytokines boosts Th1 immune responses in CD. Furthermore, anti-IL-17 therapy might affect regulatory T (Treg) cells as a cell population expressing both Th17 and Treg markers has been recently been described in CD.129 However, as RORγ deficiency in T cells caused an impressive reduction in the activity of experimental colitis,127 targeting of Th17 cells themselves rather than targeting individual Th17 cytokines might be an option for CD therapy in the future. Similarly to IL-17R signaling, targeting of IL-2R signaling in UC with the monoclonal antibody basiliximab did not result in marked improvement of disease activity, indicating that targeting of IL-2R signal transduction is not effective in this disease.130
Another interesting cytokine for IBD therapy consists of IL-6. IL-6 signaling via the sIL-6 receptor and gp130 has been shown to cause T-cell activation and mediates T-cell resistance against apoptosis in IBD patients (Figure 3).131, 132 Furthermore, antibodies against the IL-6R were effective for therapy in animal models of colitis and induced T-cell apoptosis in cell culture experiments with lamina propria cells from IBD patients. Finally, IL-6 signaling was found to be crucial for the development of colitis-associated neoplasias in mouse models,28 suggesting that targeting of IL-6 signaling might be useful for IBD therapy. Interestingly, an initial study using the humanized anti-IL-6R antibody MRA showed a clinical response in 80% of the CD patients given biweekly MRA as compared with 31% in the placebo group.133 However, no effects on mucosal healing were noted after 12 weeks. As no phase 3 studies on the use of MRA in CD were subsequently performed, the clinical efficacy of this approach needs to be confirmed by future studies. Another concept for targeting of IL-6 function in IBD consists of a recombinant gp130-Fc protein for suppression of sIL-6R signaling.134 It is expected that this approach will enter clinical testing soon.
Studies in experimental colitis have shown that additional proinflammatory cytokines might be new targets beyond TNF that could be explored for IBD therapy. Among these cytokines are IL-18, IL-21, IL-27, and IL-13.135, 136, 137, 138, 139 The targeting of IL-13 has recently gained particular attention, as blockade of IL-13 expression and function was highly effective in the treatment of experimental colitis. In addition, IL-13 is increasingly produced in UC and targets epithelial cell survival and barrier function in this disease.55, 140, 141 Current phase 2 studies will clarify whether targeting of this T-cell-derived cytokine will be beneficial in UC therapy (Figure 4).
In addition to targeting of IFN-γ and IL-17A, blockade of the CD28/B7.1-2 interactions with the monoclonal antibody abatacept was not successful for CD and UC therapy.142 Furthermore, the anti-CD3 antibody visilizumab was not effective for the treatment of corticosteroid-refractory UC.143 Similarly, blockade of the chemokine IP-10 by monoclonal antibodies (BMS936557) was not effective in a recent UC trial.144 Given these disappointing findings, new concepts for suppression of proinflammatory cytokines and T-cell activation are needed. As many studies targeting single cytokines have failed, it could be of interest to develop novel approaches for the blockade of several cytokines in IBD. Such concepts might involve small molecules for cytokine inhibition, bispecific antibodies targeting two cytokines simultaneously, Jak inhibitors or antibodies targeting cytokine subunits that affect several cytokines.145, 146 With regard to the latter possibility, antibodies against the p40 subunit of IL-12 and IL-23 have been successfully used in animal models of colitis147, 148, 149 and appear to affect Th1 and Th17 cells simultaneously in chronic intestinal inflammation.123, 150, 151 Although IL-12 favors Th1 T-cell polarization in colonic inflammation via the transcription factor STAT4, IL-23 induces STAT activation in Th17 cells and stabilizes their phenotype (Figure 2).152, 153 On the basis of promising results in phase 2 studies with anti-p40 antibodies,154, 155, 156 a recent phase 3 study has tested the efficacy of the anti-p40 antibody ustekinumab in moderate-to-severe CD (Figure 4).157 It was found that ustekinumab induces clinical responses in CD patients, especially in those previously given the anti-TNF antibody infliximab. Thus, ustekinumab may be a new treatment option for anti-TNF refractory CD.
In addition to anti-p40, recent studies have looked at the effects of Jak inhibition in IBD. Interestingly, a recent phase 2 study on the use of the Jak1/Jak3 inhibitor tofacitinib in UC showed impressive results with significantly higher response and remission rates in tofacitinib-treated patients as compared with placebo (Figure 4).158 Tofacitinib treatment also induced mucosal healing on endoscopy in many patients. If these results are confirmed in phase 3 studies, it is likely that tofacitinib will emerge as a new treatment option for UC. Tofacitinib targets both Jak1 and Jak3 and will thus block the action of numerous cytokines dependent on Jak1/Jak3, such as IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 (Figure 4).158, 159 In addition, this drug may affect the function of cytokines dependent on Jak1/Jak2, such as IL-6, IL-11, IFN-γ, and IFN-α/IFN-β. Given these pleiotropic effects on multiple cytokines, the safety profile of tofacitinib will require further analysis in future studies.
Collectively, substantial progress has been made in recent years with regard to targeting of proinflammatory cytokines. In spite of some disappointments, several agents such as ustekinumab for CD and tofacitinib for UC may find their way to the clinic and offer significant therapeutic alternatives for the management of IBD.
Inducing Treg cells and anti-inflammatory cytokines in IBD
On the basis of the finding that IL-10-deficient animals spontaneously develop T-cell-dependent colitis and colitis-associated neoplasias,160, 161 treatment with recombinant anti-inflammatory cytokines was tested for IBD therapy. Although treatment with recombinant anti-inflammatory cytokines was successful for the therapy of experimental colitis in mice, unfortunately, treatment of humans with systemic recombinant IL-10 or recombinant IL-11 did not result in improvement of clinical symptoms in CD.162, 163 Another strategy consisted of topical delivery of IL-10 to the inflamed gut by transgenic bacteria (L. lactis LL-Thy12) producing IL-10. An initial phase 1 study in 10 CD patients showed that this concept is safe but studies on clinical efficacy have not been performed.164 Another alternative could be the administration of bacterial strains that induce IL-10 production by host cells. In this context, Clostridium butyricum has been recently shown to prevent experimental colitis in mice by inducing IL-10 production of F4/80+ CD11b+ macrophages via the TLR2/MyD88 pathway.165 Collectively, these results suggested that systemic administration of the recombinant anti-inflammatory cytokines IL-10 and IL-11 might not be a good approach for the therapy of IBD. However, local administration of IL-10 could be an alternative as well as a treatment with bacterial strains that induce intestinal IL-10 production, but these approaches require further development.
Additional interesting anti-inflammatory cytokines for IBD therapy are IL-22, IL-35, and TGF-β. IL-22 induces STAT3 activation in IECs and favors IEC proliferation and colonic wound healing in vivo.25 Furthermore, IL-22 protects the mucus barrier through stimulation of mucin 1 production under intestinal inflammatory conditions and enhances the epithelial regeneration with goblet cell restitution.166 Interestingly, IL-22-producing T cells are markedly reduced in UC patients with epithelial alterations,167 suggesting that treatment with recombinant IL-22 might improve barrier function in UC by induction of STAT3-mediated proliferation in IECs and production of antimicrobial peptides. However, this concept has not been clinically tested so far. Similarly, IL-35 therapy has not been performed, although administration of recombinant IL-35 suppressed the activity of experimental colitis in mice by reducing Th1 and Th17 cytokine responses.168
The cytokine TGF-β is produced by Treg cells and suppresses T-cell-mediated colitis in animal models.169 In mice, recombinant TGF-β as well as administration of naturally occurring or inducible Treg cells have been successfully used for the treatment of experimental colitis, raising the possibility that this concept might be of relevance for treatment of IBD.170, 171, 172 With regard to Treg cells, a recent phase 1/2a study in 20 patients showed that a single injection of antigen-specific (ovalbumin) Treg cells is safe in CD and it was found that about 40% of the patients show a clinical response after treatment.173 This interesting study is consistent with the possibility that treatment with Treg cells might become available for IBD therapy, although larger phase 2/3 studies are clearly required to further develop this concept.
Treatment with recombinant TGF-β has not been considered for IBD therapy as yet, mainly because of concerns regarding the well-known fibrogenic and cancer-supporting effects of this cytokine.174 In addition, TGF-β effects in IBD T cells are inhibited by the protein Smad7 and Smad7 is markedly overexpressed in IBD patients.175 Inhibition of Smad7 via antisense DNA restored TGF-β sensitivity in IBD T cells and was effective in murine models of experimental colitis.176, 177 On the basis of these findings, a recent phase 1 study on Smad7 antisense DNA in CD patients has yielded impressive results that are currently extended in a larger phase 2 study.178 It is thus possible that targeting of TGF-β effects is a realistic option for IBD therapy in the future. Furthermore, TGF-β has been implicated in the clinical effects of worm ova therapy in IBD.179 As IBD are uncommon in underdeveloped areas of the world where many people suffer from worm infections, ingestion of live Trichuris suis ova has been tested for IBD therapy. Treatment with 2500 live ova every 3 weeks for 24 weeks was found to be effective in phase 2 studies in both CD and UC.180, 181 On the basis of these findings, larger phase 3 studies are currently under way to confirm the therapeutic efficacy of this approach. Interestingly, transcriptome analysis of T. suis predicted secreted molecules that are related to TGF-β,182 suggesting that this therapy might induce local immunosuppression by induction of Treg cells. Although further studies on this concept are needed, these findings suggest that therapy with T. suis ova might be a new option for the treatment of IBD.
Targeting cell adhesion and homing in IBD: a whole new world
Active IBD is dependent on the recruitment of mononuclear cells and leukocyte populations from the blood stream into the bowel wall. Recruitment is dependent on a series of steps known as rolling, tight binding/adhesion to endothelial cells, diapedesis, and migration of immune cells. This process is coordinated by selective adhesion molecules on the surface of immune cells and mucosal addressins on endothelial cells.183 Selective adhesion molecules include cell-surface integrins that form heterodimers by various combinations of α- and β-subunits. For gut homing of leukocytes, the interaction between α4/β7-integrins on T cells and the mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) addressin on endothelial cells appears to be of crucial relevance.
A proof-of-concept study was carried out in 1993, in which spontaneous colitis in cotton top tamarins was treated with a neutralizing antibody against the α4-integrin subunit.184 This therapy led to a significant reduction of mucosal inflammation and suggested further avenues for clinical development. Indeed, later clinical studies observed that the neutralizing humanized α4 antibody natalizumab has clinical efficacy in maintaining remission in CD, although the primary efficacy end point on induction of clinical responses at week 10 was not achieved in phase 3 studies in this disease.185 The remarkable data on maintenance of remission were associated with an improvement in quality-of-life parameters in CD. However, the use of natalizumab in CD led to rare serious adverse events caused by polyoma JC virus infection with progressive multifocal leukoencephalopathy.186 The induction of this progressive, fatal infection was probably due to the fact that natalizumab blocks α4/β1 integrins in addition to α4/β7 heterodimers and that the former integrins are important for T-cell homing to the central nervous system. Therefore, natalizumab might have caused severe immunosuppression in these patients with consecutive development of progressive multifocal leukoencephalopathy based on the blockade of the interaction between α4/β1-integrin and its ligand vascular cell adhesion molecule. To circumvent this problem, subsequent clinical trials (phase 2/3) used a second humanized monoclonal antibody, denoted vedolizumab (MLN-02/LDP-02), that specifically targets the α4/β7 heterodimer in IBD.187 Intravenous administration of vedolizumab led to significantly higher response and remission rates in UC patients as compared with placebo.188 Moreover, vedolizumab induced mucosal healing compared with placebo. Further studies showed that vedolizumab favors maintenance of remission in UC, suggesting that this substance will soon be available for clinical routine therapy in this disease. Importantly, vedolizumab also appeared to be effective for the treatment of anti-TNF refractory UC.
On the basis of these promising results, additional antiadhesion strategies are currently being tested in IBD, including the anti-β7 antibody etrolizumab.189 Etrolizumab therapy was significantly more effective for induction of remission in a recent phase 2 study in UC than placebo and further phase 3 development is expected soon (Figure 3). In addition, a MAdCAM-1 antibody targeting the α4/β7 ligand is now in clinical trials in UC. Moreover, a selective small-molecule antagonist of the CCR9 chemokine receptor that targets the interaction between CCR9 on leukocytes and its ligand CCL25 on endothelial cells is currently tested in CD of the small bowel.190 This antagonist targets a tissue-specific lymphocyte-trafficking molecule that controls T- and B-cell homing to the small intestine.191 Finally, a new concept to target lymphocyte traffic in UC consists of the sphingosine-1-phosphate receptor agonist fingolimod (FTY-720).192 This substance induces retention of leukocytes in lymph nodes and thereby prevents their entry into the gut mucosa. Clinical data will have to demonstrate whether this substance has a role in IBD therapy.
Collectively, these findings suggest that targeting of leukocyte homing is an emerging new concept for IBD therapy. It is likely that further neutralizing antibodies or small molecules for the blockade of adhesion molecules will soon enter clinical development for IBD therapy. Additional studies will have to determine the clinical relevance of this new concept in comparison to established immunosuppressive strategies including anti-TNF therapy.
Summary
In recent years, remarkable progress in IBD therapy has been made. The anti-TNF era has shown that mucosal healing is a key goal for therapy that predicts clinical remission and resection-free survival in IBD patients. Combined immunosuppression with azathioprine plus anti-TNF antibodies is a new concept for patients with severely active disease. As many patients do not respond to anti-TNF therapy, many new targets have been recently tested for induction of mucosal healing as well as for induction and maintenance of remission in IBD. It is expected that these new studies will have a marked impact on future therapy of IBD patients.
References
Danese, S & Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 365, 1713–1725 (2011).
Neurath, M.F., Finotto, S & Glimcher, L.H. The role of Th1/Th2 polarization in mucosal immunity. Nat. Med. 8, 567–573 (2002).
Baumgart, D.C. & Sandborn, W.J. Crohn’s disease. Lancet 380, 1590–1605 (2012).
Peyrin-Biroulet, L, Loftus, E.V. Jr, Colombel, J.F. & Sandborn, W.J. Long-term complications, extraintestinal manifestations, and mortality in adult Crohn’s disease in population-based cohorts. Inflamm. Bowel Dis 17, 471–478 (2011).
Bernstein, C.N., Blanchard, J.F., Kliewer, E & Wajda, A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 91, 854–862 (2001).
Foersch, S, Waldner, M.J. & Neurath, M.F. Colitis and colorectal cancer. Dig. Dis. 30, 469–476 (2012).
Hommes, D, Colombel, J.F., Emery, P, Greco, M & Sandborn, W.J. Changing Crohn’s disease management: need for new goals and indices to prevent disability and improve quality of life. J. Crohn’s Colitis 6 (Suppl 2), S224–S234 (2012).
Neurath, M.F. & Travis, S.P. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut 61, 1619–1635 (2012).
Schnitzler, F. et al. Mucosal healing predicts long-term outcome of maintenance therapy with infliximab in Crohn’s disease. Inflamm. Bowel Dis. 15, 1295–1301 (2009).
Colombel, J.F. et al. Early mucosal healing with infliximab is associated with improved long-term clinical outcomes in ulcerative colitis. Gastroenterology 141, 1194–1201 (2011).
Froslie, K.F., Jahnsen, J, Moum, B.A. & Vatn, M.H. Mucosal healing in inflammatory bowel disease: results from a Norwegian population-based cohort. Gastroenterology 133, 412–422 (2007).
Pineton de Chambrun, G, Peyrin-Biroulet, L, Lemann, M & Colombel, J.F. Clinical implications of mucosal healing for the management of IBD. Nat. Rev. Gastroenterol. Hepatol. 7, 15–29 (2010).
Rubin, D.T. We once were blind and now we see: Is it time to treat ulcerative colitis to achieve mucosal healing? Clin. Gastroenterol Hepatol 9, 456–457 (2011).
Colombel, J.F. et al. Adalimumab induces deep remission in patients with Crohn’s disease. Clin. Gastroenterol. Hepatol. in press (2013).
Krishnan, K, Arnone, B & Buchman, A. Intestinal growth factors: potential use in the treatment of inflammatory bowel disease and their role in mucosal healing. Inflamm. Bowel Dis 17, 410–422 (2011).
Paclik, D, Lohse, K, Wiedenmann, B, Dignass, A.U. & Sturm, A. Galectin-2 and -4, but not galectin-1, promote intestinal epithelial wound healing in vitro through a TGF-beta-independent mechanism. Inflamm. Bowel Dis 14, 1366–1372 (2008).
Sturm, A & Dignass, A.U. Epithelial restitution and wound healing in inflammatory bowel disease. World J. Gastroenterol 14, 348–353 (2008).
Dignass, A.U. & Podolsky, D.K. Cytokine modulation of intestinal epithelial cell restitution: central role of transforming growth factor beta. Gastroenterology 105, 1323–1332 (1993).
Dignass, A, Lynch-Devaney, K, Kindon, H, Thim, L & Podolsky, D.K. Trefoil peptides promote epithelial migration through a transforming growth factor beta-independent pathway. J. Clin. Invest. 94, 376–383 (1994).
Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 33 (Part 5), 891–895 (2005).
Moyer, R.A., Wendt, M.K., Johanesen, P.A., Turner, J.R. & Dwinell, M.B. Rho activation regulates CXCL12 chemokine stimulated actin rearrangement and restitution in model intestinal epithelia. Lab. Invest. 87, 807–817 (2007).
Vongsa, R.A., Zimmerman, N.P. & Dwinell, M.B. CCR6 regulation of the actin cytoskeleton orchestrates human beta defensin-2- and CCL20-mediated restitution of colonic epithelial cells. J. Biol. Chem. 284, 10034–10045 (2009).
Iizuka, M & Konno, S. Wound healing of intestinal epithelial cells. World J. Gastroenterol. 17, 2161–2171 (2011).
Dignass, A.U., Stow, J.L. & Babyatsky, M.W. Acute epithelial injury in the rat small intestine in vivo is associated with expanded expression of transforming growth factor alpha and beta. Gut 38, 687–693 (1996).
Pickert, G. et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 206, 1465–1472 (2009).
Nenci, A. et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561 (2007).
Grivennikov, S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 (2009).
Becker, C. et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491–501 (2004).
Seno, H, Miyoshi, H, Brown, S.L., Geske, M.J., Colonna, M & Stappenbeck, T.S. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc. Natl. Acad. Sci. USA 106, 256–261 (2009).
Goretsky, T. et al. P53 mediates TNF-induced epithelial cell apoptosis in IBD. Am. J. Pathol. 181, 1306–1315 (2012).
Gilbert, S. et al. Enterocyte STAT5 promotes mucosal wound healing via suppression of myosin light chain kinase-mediated loss of barrier function and inflammation. EMBO Mol. Med. 4, 109–124 (2012).
Han, X. et al. Regulation of intestinal barrier function by signal transducer and activator of transcription 5b. Gut 58, 49–58 (2009).
Goke, M, Kanai, M & Podolsky, D.K. Intestinal fibroblasts regulate intestinal epithelial cell proliferation via hepatocyte growth factor. Am. J. Physiol. 274 (Part 1), G809–G818 (1998).
Su, L. et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology 145, 407–415 (2013).
Bernasconi, E. et al. Granulocyte–macrophage colony-stimulating factor elicits bone marrow-derived cells that promote efficient colonic mucosal healing. Inflamm. Bowel Dis. 16, 428–441 (2010).
Huebener, P & Schwabe, R.F. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim Biophys. Acta 1832, 1005–1017 (2013).
Podolsky, D.K., Gerken, G, Eyking, A & Cario, E. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology 137, 209–220 (2009).
Ey, B, Eyking, A, Gerken, G, Podolsky, D.K. & Cario, E. TLR2 mediates gap junctional intercellular communication through connexin-43 in intestinal epithelial barrier injury. J. Biol. Chem. 284, 22332–22343 (2009).
Fukata, M. et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G1055–G1065 (2005).
Lee, J. et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat. Cell. Bio.l 8, 1327–1336 (2006).
Lee, J, Mo, J.H., Shen, C, Rucker, A.N. & Raz, E. Toll-like receptor signaling in intestinal epithelial cells contributes to colonic homoeostasis. Curr. Opin. Gastroenterol. 23, 27–31 (2007).
Rose, W.A. II, Sakamoto, K & Leifer, C.A. TLR9 is important for protection against intestinal damage and for intestinal repair. Scientific Rep. 2, 574 (2012).
Katakura, K, Lee, J, Rachmilewitz, D, Li, G, Eckmann, L & Raz, E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Invest. 115, 695–702 (2005).
Rachmilewitz, D. et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology 126, 520–528 (2004).
Rachmilewitz, D. et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology 122, 1428–1441 (2002).
Wehkamp, J. et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA 102, 18129–18134 (2005).
Ho, S.B. et al. Cysteine-rich domains of muc3 intestinal mucin promote cell migration, inhibit apoptosis, and accelerate wound healing. Gastroenterology 131, 1501–1517 (2006).
Otte, J.M. et al. Human beta defensin 2 promotes intestinal wound healing in vitro. J. Cell. Biochem. 104, 2286–2297 (2008).
Pickard, K.M., Bremner, A.R., Gordon, J.N. & MacDonald, T.T. Microbial–gut interactions in health and disease. Immune responses. Best Pract. Res. Clin. Gastroenterol. 18, 271–285 (2004).
Hering, N.A., Fromm, M & Schulzke, J.D. Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J. Physiol. 590 (Part 5), 1035–1044 (2012).
McGuckin, M.A., Eri, R, Simms, L.A., Florin, T.H. & Radford-Smith, G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm. Bowel Dis. 15, 100–113 (2009).
John, L.J., Fromm, M & Schulzke, J.D. Epithelial barriers in intestinal inflammation. Antioxidants Redox Signal. 15, 1255–1270 (2011).
Fischer, A, Gluth, M, Pape, U.F., Wiedenmann, B, Theuring, F & Baumgart, D.C. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-alpha on tight junction proteins and signaling pathways in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G970–G979 (2013).
Zeissig, S. et al. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut 53, 1295–1302 (2004).
Heller, F. et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129, 550–564 (2005).
Boltin, D, Perets, T.T., Vilkin, A & Niv, Y. Mucin function in inflammatory bowel disease: an update. J. Clin. Gastroenterol. 47, 106–111 (2013).
Sheng, Y.H., Hasnain, S.Z., Florin, T.H. & McGuckin, M.A. Mucins in inflammatory bowel diseases and colorectal cancer. J. Gastroenterol. Hepatol. 27, 28–38 (2012).
Johansson, M.E., Larsson, J.M. & Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host–microbial interactions. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4659–4665 (2011).
Hansson, G.C. & Johansson, M.E. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut Microbes 1, 51–54 (2010).
Dorofeyev, A.E., Vasilenko, I.V., Rassokhina, O.A. & Kondratiuk, R.B. Mucosal barrier in ulcerative colitis and Crohn’s disease. Gastroenterol. Res. Pract. 2013, 431231 (2013).
Stremmel, W, Hanemann, A, Ehehalt, R, Karner, M & Braun, A. Phosphatidylcholine (lecithin) and the mucus layer: evidence of therapeutic efficacy in ulcerative colitis? Dig. Dis. 28, 490–496 (2010).
Stremmel, W, Merle, U, Zahn, A, Autschbach, F, Hinz, U & Ehehalt, R. Retarded release phosphatidylcholine benefits patients with chronic active ulcerative colitis. Gut 54, 966–971 (2005).
Gunther, C. et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011).
Salzman, N.H., Ghosh, D, Huttner, K.M., Paterson, Y & Bevins, C.L. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 422, 522–526 (2003).
Kruis, W. et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 53, 1617–1623 (2004).
Gersemann, M, Wehkamp, J & Stange, E.F. Innate immune dysfunction in inflammatory bowel disease. J. Intern. Med. 271, 421–428 (2012).
Hofmann, C. et al. T cell-dependent protective effects of CpG motifs of bacterial DNA in experimental colitis are mediated by CD11c+ dendritic cells. Gut 59, 1347–1354 (2010).
Musch, E. et al. Topical treatment with the Toll-like receptor agonist DIMS0150 has potential for lasting relief of symptoms in patients with chronic active ulcerative colitis by restoring glucocorticoid sensitivity. Inflamm. Bowel Dis. 19, 283–292 (2013).
Prantera, C, Lochs, H, Grimaldi, M, Danese, S, Scribano, M.L. & Gionchetti, P. Rifaximin-extended intestinal release induces remission in patients with moderately active Crohn’s disease. Gastroenterology 142, 473–481 e474 (2012).
Prantera, C. et al. Antibiotic treatment of Crohn’s disease: results of a multicentre, double blind, randomized, placebo-controlled trial with rifaximin. Aliment. Pharmacol. Therap. 23, 1117–1125 (2006).
Papi, C, Spurio, F.F., Margagnoni, G & Aratari, A. Randomized controlled trials in prevention of postsurgical recurrence in Crohn’s disease. Rev. Recent Clin. Trials 7, 307–313 (2012).
Gardet, A & Xavier, R.J. Common alleles that influence autophagy and the risk for inflammatory bowel disease. Curr. Opin. Immunol. 24, 522–529 (2012).
Kuballa, P, Nolte, W.M., Castoreno, A.B. & Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 30, 611–646 (2012).
Chassaing, B. et al. Crohn disease-associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J. Clin. Invest. 121, 966–975 (2011).
Scharl, M & Rogler, G. Inflammatory bowel disease: dysfunction of autophagy? Dig. Dis. 30 (Suppl 3), 12–19 (2012).
Bringer, M.A., Billard, E, Glasser, A.L., Colombel, J.F. & Darfeuille-Michaud, A. Replication of Crohn’s disease-associated AIEC within macrophages is dependent on TNF-alpha secretion. Lab. Invest. 92, 411–419 (2012).
Flammer, J.R. & Rogatsky, I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol. Endocrinol. 25, 1075–1086 (2011).
Kornbluth, A, Present, D.H., Lichtiger, S & Hanauer, S. Cyclosporin for severe ulcerative colitis: a user’s guide. Am. J. Gastroenterol. 92, 1424–1428 (1997).
Lichtiger, S & Present, D.H. Preliminary report: cyclosporin in treatment of severe active ulcerative colitis. Lancet 336, 16–19 (1990).
Feagan, B.G. et al. A comparison of methotrexate with placebo for the maintenance of remission in Crohn's disease. North American Crohn’s Study Group Investigators. N. Engl. J. Med. 342, 1627–1632 (2000).
Feagan, B.G. et al. Methotrexate for the treatment of Crohn’s disease. The North American Crohn’s Study Group Investigators. N.Engl. J. Med. 332, 292–297 (1995).
Neurath, M.F. et al. Methotrexate specifically modulates cytokine production by T cells and macrophages in murine collagen-induced arthritis (CIA): a mechanism for methotrexate-mediated immunosuppression. Clin. Exp. Immunol. 115, 42–55 (1999).
Hildner, K. et al. Tumour necrosis factor (TNF) production by T cell receptor-primed T lymphocytes is a target for low dose methotrexate in rheumatoid arthritis. Clin. Exp. Immunol. 118, 137–146 (1999).
Kaminuma, O. Selective inhibitors of nuclear factor of activated T cells: potential therapeutic drugs for the treatment of immunological and inflammatory diseases. Inflamm. Allergy DrugTargets 7, 35–40 (2008).
Fric, J, Zelante, T, Wong, A.Y., Mertes, A, Yu, H.B. & Ricciardi-Castagnoli, P. NFAT control of innate immunity. Blood 120, 1380–1389 (2012).
Daniel, C, Gerlach, K, Vath, M, Neurath, M.F. & Weigmann, B. Nuclear factor of activated T cells—a transcription factor family as critical regulator in lung and colon cancer. Int. J. Cancer (in press) (2013).
Weigmann, B & Neurath, M.F. Selective targeting of activated T cells in chronic intestinal inflammation. Gut 58, 747–748 (2009).
Tiede, I. et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Invest. 111, 1133–1145 (2003).
Poppe, D. et al. Azathioprine suppresses ezrin-radixin-moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins. J. Immunol. 176, 640–651 (2006).
Scheller, J, Chalaris, A, Garbers, C & Rose-John, S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 32, 380–387 (2011).
Wajant, H, Pfizenmaier, K & Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 10, 45–65 (2003).
Colombel, J.F. et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N. Engl J. Med. 362, 1383–1395 (2010).
Colombel, J.F. et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 132, 52–65 (2007).
Hanauer, S.B. et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 130, 323–333 quiz 591 (2006).
Evans, A.T. & Lee, S.D. A review and expert opinion of the use of certolizumab for Crohn’s disease. Expert Opin. Biol. Ther. 12, 363–370 (2012).
Rutgeerts, P, Vermeire, S & Van Assche, G. Predicting the response to infliximab from trough serum levels. Gut 59, 7–8 (2010).
Present, D.H. Review article: the efficacy of infliximab in Crohn’s disease—healing of fistulae. Aliment. Pharmacol. Therap. 13 (Suppl 4), 23–28 discussion 38 (1999).
Hanauer, S.B. et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 359, 1541–1549 (2002).
Sandborn, W.J. et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology (in press) (2013).
Sandborn, W.J. et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology (in press) (2013).
Gecse, K.B. et al. Biosimilars in IBD: hope or expectation? Gut 62, 803–807 (2013).
Vandenbroucke, K. et al. Orally administered L. lactis secreting an anti-TNF Nanobody demonstrate efficacy in chronic colitis. Mucosal Immunol. 3, 49–56 (2010).
Assier, E. et al. Modulation of anti-tumor necrosis factor alpha (TNF-alpha) antibody secretion in mice immunized with TNF-alpha kinoid. Clin. Vaccine Immunol. 19, 699–703 (2012).
Danese, S, Colombel, J.F., Peyrin-Biroulet, L, Rutgeerts, P & Reinisch, W. Review article: the role of anti-TNF in the management of ulcerative colitis—past, present and future. Aliment. Pharmacol. Therap. 37, 855–866 (2013).
Danese, S, Fiorino, G & Reinisch, W. Review article: causative factors and the clinical management of patients with Crohn’s disease who lose response to anti-TNF-alpha therapy. Aliment. Pharmacol. Therap. 34, 1–10 (2011).
Sandborn, W.J. & Hanauer, S.B. Antitumor necrosis factor therapy for inflammatory bowel disease: a review of agents, pharmacology, clinical results, and safety. Inflamm. Bowel Dis. 5, 119–133 (1999).
Colombel, J.F. et al. Adalimumab safety in global clinical trials of patients with Crohn’s disease. Inflamm. Bowel Dis. 15, 1308–1319 (2009).
Eser, A, Primas, C & Reinisch, W. Drug monitoring of biologics in inflammatory bowel disease. Curr. Opin. Gastroenterol. 29, 391–396 (2013).
Vos, A.C., Wildenberg, M.E., Duijvestein, M, Verhaar, A.P., van den Brink, G.R. & Hommes, D.W. Anti-tumor necrosis factor-alpha antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology 140, 221–230 (2011).
Vos, A.C. et al. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm. Bowel Dis. 18, 401–408 (2012).
Van den Brande, J.M. et al. Prediction of antitumour necrosis factor clinical efficacy by real-time visualisation of apoptosis in patients with Crohn’s disease. Gut 56, 509–517 (2007).
Atreya, R. et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14(+) macrophages. Gastroenterology 141, 2026–2038 (2011).
Mitoma, H. et al. Infliximab induces potent anti-inflammatory responses by outside-to-inside signals through transmembrane TNF-alpha. Gastroenterology 128, 376–392 (2005).
Van den Brande, J.M. et al. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology 124, 1774–1785 (2003).
Sandborn, W.J. et al. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology 121, 1088–1094 (2001).
Atreya, R. et al. Prediction of anti-TNF antibody responses in crohn's disease by endoscopic molecular imaging in vivo. Gastroenterology 142 (Suppl), S535 (2012).
Perrier, C. et al. Neutralization of membrane TNF, but not soluble TNF, is crucial for the treatment of experimental colitis. Inflamm. Bowel Dis. 19, 246–253 (2013).
Lethaby, A, Lopez-Olivo, M.A., Maxwell, L, Burls, A, Tugwell, P & Wells, G.A. Etanercept for the treatment of rheumatoid arthritis. Cochrane Database Syst. Rev. 5, CD004525 (2013).
Papp, K.A. et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 366, 1181–1189 (2012).
Plushner, S.L. Tocilizumab: an interleukin-6 receptor inhibitor for the treatment of rheumatoid arthritis. Ann. Pharmacother. 42, 1660–1668 (2008).
Fuss, I.J. et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 157, 1261–1270 (1996).
Monteleone, I, Pallone, F & Monteleone, G. Th17-related cytokines: new players in the control of chronic intestinal inflammation. BMC Med. 9, 122 (2011).
De Nitto, D, Sarra, M, Cupi, M.L., Pallone, F & Monteleone, G. Targeting IL-23 and Th17-cytokines in inflammatory bowel diseases. Curr. Pharm. Des. 16, 3656–3660 (2010).
Kleinschek, M.A. et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 206, 525–534 (2009).
Reinisch, W. et al. Fontolizumab in moderate to severe Crohn’s disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm. Bowel Dis. 16, 233–242 (2010).
Reinisch, W. et al. A dose escalating, placebo controlled, double blind, single dose and multidose, safety and tolerability study of fontolizumab, a humanised anti-interferon gamma antibody, in patients with moderate to severe Crohn’s disease. Gut 55, 1138–1144 (2006).
Leppkes, M. et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136, 257–267 (2009).
Hueber, W. et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693–1700 (2012).
Hovhannisyan, Z, Treatman, J, Littman, D.R. & Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 140, 957–965 (2011).
Sands, B.E. et al. Basiliximab does not increase efficacy of corticosteroids in patients with steroid-refractory ulcerative colitis. Gastroenterology 143, 356–364 e351 (2012).
Atreya, R & Neurath, M.F. Signaling molecules: the pathogenic role of the IL-6/STAT-3 trans signaling pathway in intestinal inflammation and in colonic cancer. Curr. Drug Targets 9, 369–374 (2008).
Atreya, R. et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 6, 583–588 (2000).
Ito, H. et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 126, 989–996 discussion 947 (2004).
Jones, S.A., Scheller, J & Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Invest. 121, 3375–3383 (2011).
Siegmund, B. et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R1264–R1273 (2001).
Stolfi, C. et al. Involvement of interleukin-21 in the regulation of colitis-associated colon cancer. J. Exp. Med. 208, 2279–2290 (2011).
Cox, J.H., Kljavin, N.M., Ramamoorthi, N, Diehl, L, Batten, M & Ghilardi, N. IL-27 promotes T cell-dependent colitis through multiple mechanisms. J. Exp. Med. 208, 115–123 (2011).
Heller, F, Fuss, I.J., Nieuwenhuis, E.E., Blumberg, R.S. & Strober, W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 17, 629–638 (2002).
Kawashima, R. et al. Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice—a pathway associated with ulcerative colitis. Gastroenterology 141, 2119–2129 e2118 (2011).
Fuss, I.J. & Strober, W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol 1 (Suppl 1), S31–S33 (2008).
Fuss, I.J. et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Invest. 113, 1490–1497 (2004).
Sandborn, W.J. et al. Abatacept for Crohn’s disease and ulcerative colitis. Gastroenterology 143, 62–69 e64 (2012).
Sandborn, W.J. et al. Anti-CD3 antibody visilizumab is not effective in patients with intravenous corticosteroid-refractory ulcerative colitis. Gut 59, 1485–1492 (2010).
Mayer, L. et al. Anti-IP-10 antibody (BMS-936557) for ulcerative colitis: a phase II randomised study. Gut (in press) (2013).
Dotan, I. et al. A randomised placebo-controlled multicentre trial of intravenous semapimod HCl for moderate to severe Crohn’s disease. Gut 59, 760–766 (2010).
Fournier, P & Schirrmacher, V. Bispecific antibodies and trispecific immunocytokines for targeting the immune system against cancer: preparing for the future. BioDrugs 27, 35–53 (2013).
Neurath, M.F., Fuss, I, Kelsall, B.L., Stuber, E & Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182, 1281–1290 (1995).
Davidson, N.J., Hudak, S.A., Lesley, R.E., Menon, S, Leach, M.W. & Rennick, D.M. IL-12 but not IFN-gamma, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J. Immunol. 161, 3143–3149 (1998).
Fuss, I.J., Marth, T, Neurath, M.F., Pearlstein, G.R., Jain, A & Strober, W. Anti-interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in mice. Gastroenterology 117, 1078–1088 (1999).
Hue, S. et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483 (2006).
Monteleone, I, Sarra, M, Pallone, F & Monteleone, G. Th17-related cytokines in inflammatory bowel diseases: friends or foes? Curr. Mol. Med. 12, 592–597 (2012).
Valatas, V, He, J, Rivollier, A, Kolios, G, Kitamura, K & Kelsall, B.L. Host-dependent control of early regulatory and effector T-cell differentiation underlies the genetic susceptibility of RAG2-deficient mouse strains to transfer colitis. Mucosal Immunol. 6, 601–611 (2013).
Wirtz, S. et al. Cutting edge: chronic intestinal inflammation in STAT-4 transgenic mice: characterization of disease and adoptive transfer by TNF- plus IFN-gamma-producing CD4+ T cells that respond to bacterial antigens. J. Immunol. 162, 1884–1888 (1999).
Fuss, I.J. et al. Both IL-12p70 and IL-23 are synthesized during active Crohn’s disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 12, 9–15 (2006).
Mannon, P.J. et al. Anti-interleukin-12 antibody for active Crohn’s disease. N. Engl. J. Med. 351, 2069–2079 (2004).
Sandborn, W.J. et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology 135, 1130–1141 (2008).
Sandborn, W.J. et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 367, 1519–1528 (2012).
Sandborn, W.J. et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 367, 616–624 (2012).
Liao, W, Lin, J.X. & Leonard, W.J. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 23, 598–604 (2011).
Davidson, N.J. et al. T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J. Exp. Med. 184, 241–251 (1996).
Erdman, S.E. et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 63, 6042–6050 (2003).
Fedorak, R.N. et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology 119, 1473–1482 (2000).
Herrlinger, K.R. et al. Randomized, double blind controlled trial of subcutaneous recombinant human interleukin-11 versus prednisolone in active Crohn’s disease. Am. J. Gastroenterol. 101, 793–797 (2006).
Braat, H. et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 4, 754–759 (2006).
Hayashi, A. et al. A single strain of Clostridium butyricum induces intestinal IL-10-producing macrophages to suppress acute experimental colitis in mice. Cell Host Microbe 13, 711–722 (2013).
Mizoguchi, A. Healing of intestinal inflammation by IL-22. Inflamm. Bowel Dis. 18, 1777–1784 (2012).
Leung, J.M. et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. (in press) (2013).
Wirtz, S, Billmeier, U, McHedlidze, T, Blumberg, R.S. & Neurath, M.F. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 141, 1875–1886 (2011).
Fahlen, L. et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J. Exp. Med. 201, 737–746 (2005).
Uhlig, H.H. et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 177, 5852–5860 (2006).
Izcue, A, Coombes, J.L. & Powrie, F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 212, 256–271 (2006).
Fantini, M.C. et al. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 55, 671–680 (2006).
Desreumaux, P. et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 143, 1207–1217 e1201–1202 (2012).
Shi, Y & Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700 (2003).
Monteleone, G, Boirivant, M, Pallone, F & Mac Donald, T.T. TGF-beta1 and Smad7 in the regulation of IBD. Mucosal Immunol. 1 (Suppl 1), S50–S53 (2008).
Boirivant, M. et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology 131, 1786–1798 (2006).
Fantini, M.C. et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology 136, 1308–1316 e1301–1303 (2009).
Monteleone, G. et al. Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn's disease. Mol. Ther. 20, 870–876 (2012).
Weinstock, J.V. Autoimmunity: the worm returns. Nature 491, 183–185 (2012).
Summers, R.W., Elliott, D.E., Urban, J.F. Jr., Thompson, R.A. & Weinstock, J.V. Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology 128, 825–832 (2005).
Summers, R.W., Elliott, D.E., Urban, J.F. Jr., Thompson, R & Weinstock, J.V. Trichuris suis therapy in Crohn’s disease. Gut 54, 87–90 (2005).
Cantacessi, C. et al. The transcriptome of Trichuris suis—first molecular insights into a parasite with curative properties for key immune diseases of humans. PLoS One 6, e23590 (2011).
Podolsky, D.K. Selective adhesion-molecule therapy and inflammatory bowel disease—a tale of Janus? N. Engl. J. Med. 353, 1965–1968 (2005).
Podolsky, D.K. et al. Attenuation of colitis in the cotton-top tamarin by anti-alpha 4 integrin monoclonal antibody. J Clin Invest 92, 372–380 (1993).
Sandborn, W.J. et al. Natalizumab induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 353, 1912–1925 (2005).
Van Assche, G. et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N. Engl. J. Med. 353, 362–368 (2005).
Gledhill, T & Bodger, K. New and emerging treatments for ulcerative colitis: a focus on vedolizumab. Biologics 7, 123–130 (2013).
Parikh, A. et al. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm. Bowel Dis. 18, 1470–1479 (2012).
Rutgeerts, P.J. et al. A randomised phase I study of etrolizumab (rhuMAb beta7) in moderate to severe ulcerative colitis. Gut 62, 1122–1130 (2012).
Thomas, S & Baumgart, D.C. Targeting leukocyte migration and adhesion in Crohn's disease and ulcerative colitis. Inflammopharmacology 20, 1–18 (2012).
Papadakis, K.A. et al. CC chemokine receptor 9 expression defines a subset of peripheral blood lymphocytes with mucosal T cell phenotype and Th1 or T-regulatory 1 cytokine profile. J. Immunol. 171, 159–165 (2003).
Bamias, G, Clark, D.J. & Rivera-Nieves, J. Leukocyte traffic blockade as a therapeutic strategy in inflammatory bowel disease. Curr. Drug Targets advance online publication, 25 April 2013 (e-pub ahead of print).
Neurath, M.F. et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J. Exp. Med. 195, 1129–1143 (2002).
Kobayashi, T. et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut 57, 1682–1689 (2008).
Acknowledgements
The research of M.F.N. is supported by the German Research Council DFG, the DFG Schwerpunktprogramm Microbiota, the KFO CEDER, the German Cancer Aid, Comprehensive Cancer Center CCC-EMN, and the IZKF Erlangen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
M.F.N. has served as an advisor to MSD, Pentax, Giuliani, 4SC AG, Teva, and Abbott.
Rights and permissions
About this article
Cite this article
Neurath, M. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol 7, 6–19 (2014). https://doi.org/10.1038/mi.2013.73
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2013.73
This article is cited by
-
Critical role of interferons in gastrointestinal injury repair
Nature Communications (2021)
-
Upregulation of complement C1q reflects mucosal regeneration in a mouse model of colitis
Medical Molecular Morphology (2021)
-
Corticosteroid enhances epithelial barrier function in intestinal organoids derived from patients with Crohn’s disease
Journal of Molecular Medicine (2021)
-
Activation of mTORC1 by LSECtin in macrophages directs intestinal repair in inflammatory bowel disease
Cell Death & Disease (2020)
-
Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD
Nature Reviews Gastroenterology & Hepatology (2019)
